


Introducción
La aparición de insuficiencia respiratoria en un lactante nacido a término durante la primera semana de vida es una situación excepcional. Entre las causas cabe considerar infecciones, cardiopatías congénitas, síndrome por aspiración de meconio, hipertensión pulmonar persistente, malformaciones torácicas y/o pulmonares o enfermedades pulmonares difusas. Estas últimas incluyen un grupo heterogéneo de patologías, la mayoría de las cuales son idiopáticas, caracterizadas por infiltrados pulmonares difusos, alteraciones funcionales de tipo restrictivo y del intercambio gaseoso1. Algunas enfermedades del intersticio pulmonar (EIP) son específicas de lactantes y niños pequeños, como la neumonitis intersticial celular de la infancia, conocida también como glicogenosis intersticial pulmonar, la neumonitis crónica de la infancia y la taquipnea persistente del lactante asociada con hiperplasia de células neuroendocrinas1. Otras formas de EIP pueden afectar a lactantes, niños y adultos jóvenes. Éstas incluyen la deficiente o anómala expresión de las proteínas B y C (SP-B y SP-C) del surfactante, donde el proceso inflamatorio que lleva a la EIP se inicia por un daño a las estructuras alveolares e intersticiales debido a la acumulación de material proteináceo en el espacio alveolar o en los neumocitos (alveolitis)2-12.
El surfactante pulmonar se compone de lípidos y proteínas específicas sintetizadas y secretadas en el lumen por células alveolares especializadas (neumocitos tipo II). Su función es disminuir la tensión superficial del alvéolo y prevenir la atelectasia al final de la espiración. Entre las proteínas del surfactante, la SP-C es la más hidrófoba. Su forma alveolar es un péptido de 34-35 aminoácidos, obtenido por proteólisis de un precursor de 21 kDa. La SP-C es codificada por el gen SFTPC localizado en el cromosoma 8 y su secuencia aminoacídica está muy conservada a través de las especies. Presenta un dominio en α-hélice lipídico y residuos de ácido palmítico en posición 5 y 6 que justifican su fuerte asociación con los fosfolípidos del surfactante13. La SP-C madura se conserva en los cuerpos lamelares de los neumocitos tipo II antes de ser secretada en la luz alveolar. La SP-C parece mejorar la distribución y la estabilidad de los fosfolípidos y es un componente fundamental de varios preparados de surfactante mamífero utilizado para tratar el síndrome de insuficiencia respiratoria aguda del pretérmino.
Existen estudios recientes que han asociado la producción deficiente de SP-C, o mutaciones en el gen que la codifica, con enfermedades pulmonares graves de distribución familiar4-6. Se presenta una nueva mutación dominante en el gen SFTPC asociada con EIP en tres personas de una familia española y la evolución favorable de una de ellas tras practicarse trasplante pulmonar.
Observación clínica
Paciente 1
Lactante de sexo femenino, nacida a la semana 39 de edad gestacional y peso al nacimiento de 3.070 g que presentaba taquipnea con aleteo nasal desde la primera semana de vida. Se trataba de la tercera hija de padres no consanguíneos; el embarazo y el parto transcurrieron sin incidencias. No se observaron historia de tos, fiebre, infección o exposición ambiental a tóxicos en la anamnesis o la exploración física. Tampoco se observaban signos de enfermedades cardíacas, dermatológicas, gastrointestinales, hepáticas, inmunológicas o renales, ni alteraciones esqueléticas. Inicialmente no se observó cianosis, ni con las tomas ni con el llanto. Al ingreso presentaba una oxigenación capilar por pulsioximetría de 80 % (sin oxígeno). A partir del segundo mes de vida su curva de crecimiento ponderal se mantuvo por debajo del percentil 3, a pesar de un apetito normal y una alimentación correcta. La situación respiratoria empeoraba de manera progresiva y a pesar del tratamiento con corticoides sistémicos y azatioprina, la mejoría fue sólo transitoria. A la edad de 6 meses, tras lavado broncoalveolar y biopsia pulmonar, fue diagnosticada de proteinosis alveolar con moderada fibrosis intersticial. A los 9 meses de edad entró en un programa de trasplante pulmonar y fue trasplantada a la edad de 13 meses. En la actualidad la niña tiene 5 años y presenta una actividad física normal.
Paciente 2
Es el hermano de la paciente 1. Presentó insuficiencia respiratoria desde el período neonatal y se le diagnosticó a los 4 meses de vida, por biopsia pulmonar, una neumonitis intersticial aguda y difusa. Fue tratado inicialmente con corticoides sistémicos; posteriormente se probaron azatioprina, ciclosporina y ciclofosfamida sin que se observara mejoría en su función pulmonar, ni disminución en las necesidades de oxígeno. Falleció por insuficiencia respiratoria a la edad de 3 años y 10 meses, 2 años y 3 meses antes del nacimiento de la paciente 1. La autopsia mostró alteraciones pulmonares compatibles con EIP.
Paciente 3
Es un varón de 37 años, padre de los pacientes 2 y 1. Presentaba insuficiencia respiratoria moderada-grave y acropaquias desde la infancia. Se le diagnosticó fibrosis pulmonar idiopática a la edad de 26 años, por tomografía computarizada (TC), pruebas de función pulmonar y lavado broncoalveolar. Se trató con corticoides sistémicos, con mejoría clínica y de la capacidad vital máxima, del volumen espiratorio en el primer segundo y de la capacidad de difusión del monóxido de carbono. El paciente no quiso someterse a biopsia pulmonar ni a ulteriores tratamientos.
Se obtuvieron muestras de tejido pulmonar de los pacientes 1 y 2, y muestra de sangre periférica de los pacientes 1 y 3. Las muestras se recogieron como parte de un programa que estudiaba lactantes con enfermedad pulmonar de origen desconocido en busca de mutaciones en los genes de las proteínas del surfactante. Las comisiones éticas de los centros participantes en el estudio aprobaron el protocolo y el consentimiento informado escrito para los estudios genéticos fue firmado por la madre de los pacientes 1 y 2.
El análisis anatomopatológico e inmunohistoquímico, realizado como se ha descrito anteriormente4,5 sobre tejidos de la biopsia pulmonar del paciente 1 cuando tenía 6 meses de vida, mostró fibrosis intersticial, espesamiento alveolar con muscularización de los septos alveolares, hiperplasia del epitelio alveolar y proteinosis alveolar con macrófagos grandes y espumosos. Los estudios del tejido pulmonar obtenido en el momento de la autopsia del paciente 2 mostraron neumonitis intersticial difusa, con fibrosis, inflamación crónica, hipertensión pulmonar y bronquiectasias.
El estudio del tejido pulmonar con anticuerpos específicos para la forma madura de SP-B, su precursor y SP-A mostraron una distribución normal de estas proteínas, mientras que el precursor de SP-C se encontraba solamente en las células tipo II del epitelio alveolar y no en el material proteináceo contenido en los espacios alveolares (fig. 1). En la preparación no se encontró forma madura de SP-C (datos no mostrados).
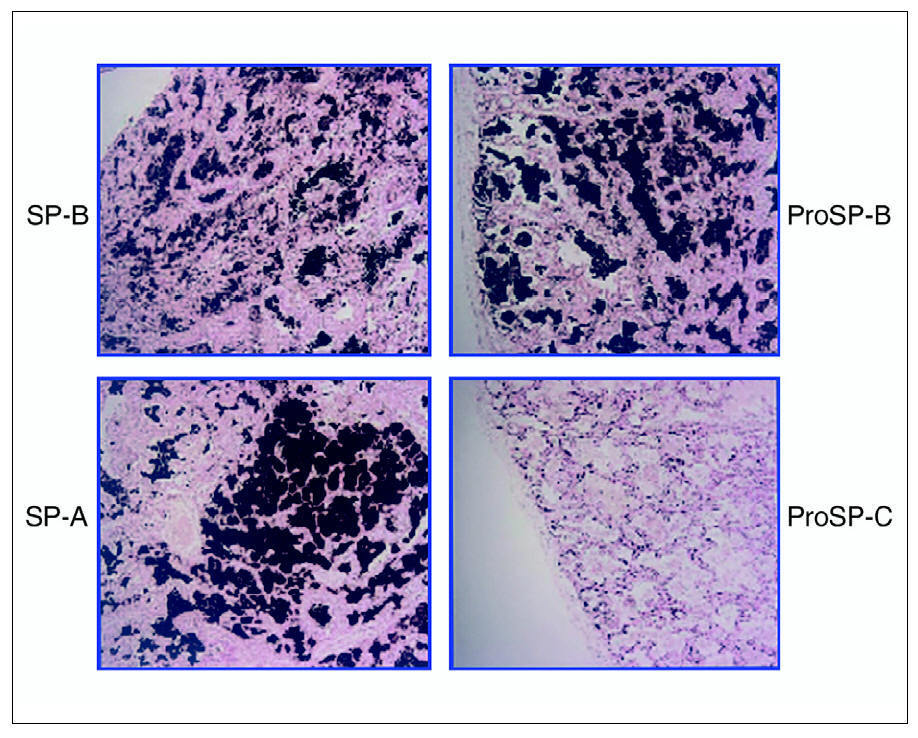
Figura 1. Inmunohistoquímica para la detección de las proteínas del surfactante, realizada sobre material de la biopsia pulmonar de la paciente 1, obtenida en el momento del diagnóstico a la edad de 6 meses. Se pueden observar la distribución de la proteína B del surfactante (SP-B) y de su precursor (proSP-B) en los espacios alveolares, así como la presencia de la proteína A del surfactante (SP-A) en la misma localización. El material contenido en los alvéolos es negativo para la determinación del precursor de la proSP-C.
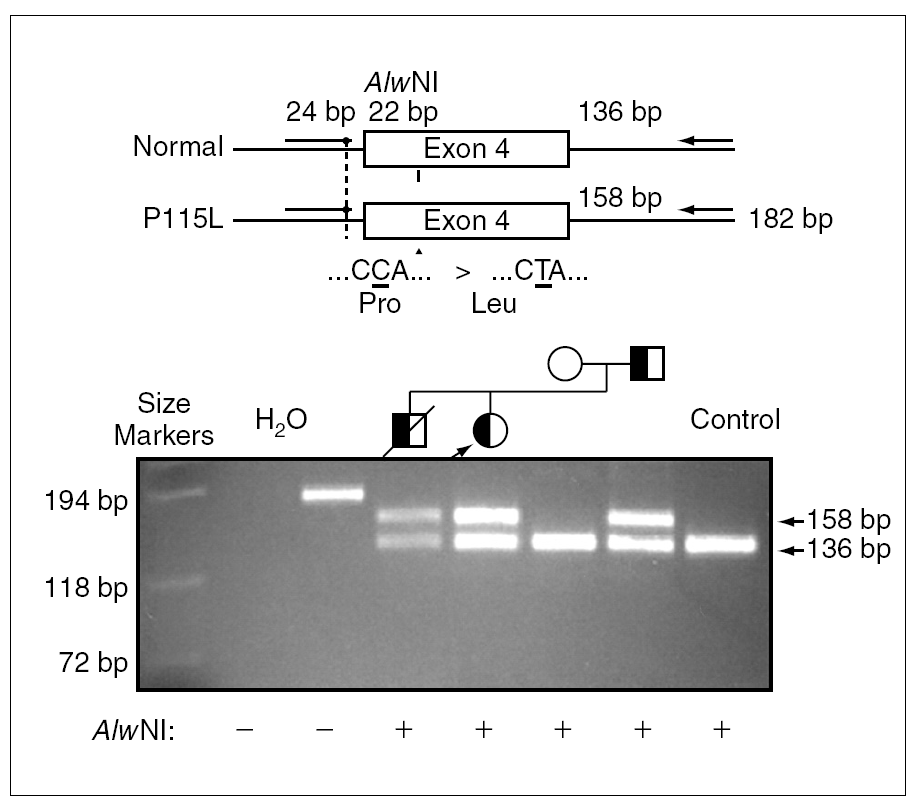
Debido a la distribución anómala del precursor de SP-C se secuenció el gen correspondiente del paciente 1 y se detectó una mutación puntual en el exón 4 de un alelo, donde la C en posición 2224 (GenBanK® número de acceso J0389014) estaba sustituida por una T (fig. 2), con pérdida de un sitio de restricción para la enzima AlwNI. En la proteína esta sustitución produce un cambio de prolina a leucina en el codón 115. La misma mutación P115L en heterozigosis se encontró también en los pacientes 2 y 3, y no se detectó ni en la madre ni en 50 individuos controles (100 cromosomas) pertenecientes a la misma población (fig. 3).
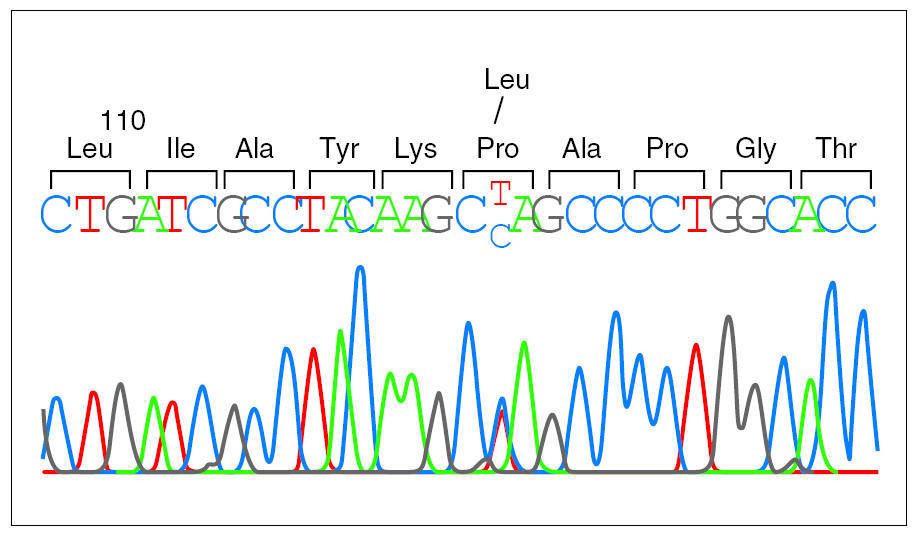
Figura 2.Secuencia del gen de la proteína C del surfactante en el paciente 1. Se puede observar la sustitución de una base en el codón 115, donde un alelo presenta una citosina y el otro una timina, cuya coincidencia se identifica con las letras C y T en la secuencia presentada.
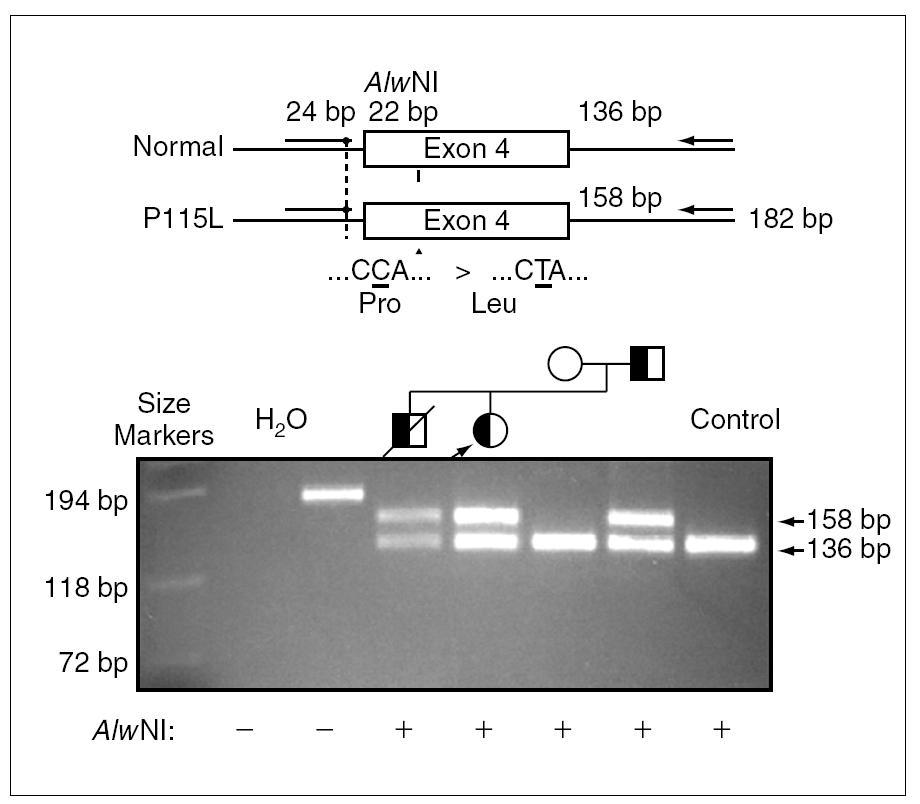
Figura 3.La sustitución de una C por una T causa la pérdida de un sitio de restricción para la enzima AlwNI. Productos de reacción en cadena de la polimerasa (PCR) en ADN genómico amplificados por medio de primers externos al exón 4 del gen de la proteína C del surfactante y digeridos con la enzima de restricción AlwNI, se han separado por electroforesis en un gel de agarosa. La paciente 1 (calle 5) presenta dos fragmentos correspondientes al alelo digerido, el normal y al alelo mutado, que no se puede digerir. Se encontró la misma mutación en heterozigosis también en los pacientes 2 y 3 (calle 4 y 7), y no se detectó ni en la madre (calle 6) ni en un control (calle 8).
Discusión
El surfactante pulmonar es una mezcla de lípidos y proteínas necesaria para reducir la tensión superficial y prevenir la atelectasia al final de la espiración. La deficiencia de surfactante pulmonar es la causa principal del síndrome de insuficiencia respiratoria aguda del recién nacido pretérmino. La SP-C es la proteína más hidrófoba del surfactante y está presente en las preparaciones purificadas de surfactante utilizadas en la práctica clínica. En los últimos años se ha clarificado la función de SP-C, y se han asociado las mutaciones en esta proteína con la EIP4-11. Se ha identificado una mutación en el aminoácido prolina correspondiente al codón 115, un residuo altamente conservado en la evolución (en dicha posición se ha encontrado una prolina en todas las especies de mamífero en las cuales se conoce la secuencia de la SP-C). El hecho de que en la familia presentada la mutación P115L sea responsable de la enfermedad se apoya en el dato de que todos los individuos afectados llevan la P115L, mientras que no se ha encontrado en ninguno de los sujetos controles y asintomáticos. La mutación P115L implica una sustitución de un aminoácido neutro altamente conservado por un aminoácido no polar que se localiza en el dominio carboxiterminal de la SP-C, una región crucial para el correcto plegamiento y procesamiento del precursor de la SP-C7,11,13. Los ratones modificados genéticamente mediante recombinación homóloga, que son deficientes en la forma madura de la SP-C, muestran una estabilidad alterada del surfactante pulmonar, y el fenotipo se hace más evidente con el envejecimiento, cuando acaban por presentar una enfermedad pulmonar grave con enfisema, infiltrado monocitario, displasia de células epiteliales y acumulación intracelular atípica de lípidos en neumocitos tipo II y macrófagos alveolares, hallazgos histológicos compatibles con neumonitis intersticial15,16. Además, se ha observado la formación de agregados y la alteración del transporte intracelular del SP-C normal mediante experimentos in vitro en los que se produce una deleción del exón 4 del gen SP-C humano, así como en los mutantes del gen de rata sin el dominio aminoterminal, funcionando en ambos casos como un dominante negativo17 mientras que la expresión de SP-C humana mutada en ratones transgénicos es letal por impedir la formación del pulmón durante el período embrionario18. Estas observaciones pueden explicar por qué in vivo el alelo normal no puede suplir el alelo anormal y no se encuentra SP-C madura y funcionante en el pulmón de los pacientes descritos. La expresión y el procesamiento del SP-C se regulan de manera evolutiva, así que el desarrollo posnatal de la enfermedad pulmonar puede deberse a la progresiva producción y/o acumulación del precursor anómalo de SP-C, con la consecuente aparición de daño celular, inflamación crónica intersticial y fibrosis pulmonar. La presencia de cuerpos de inclusión es per se causa suficiente de enfermedad, por ejemplo en el hígado de pacientes con déficit de a1-antitripsina (especialmente el fenotipo ZZ), o en el cerebro en algunos tipos de demencia19,20. Fenómenos como la penetración incompleta, la expresión variable, la anticipación clínica o la implicación de otros genes pueden explicar por qué los síntomas son más leves en el padre.
Agradecimientos
A los Dres. L. Nogee, del Departamento de Pediatría de la Facultad de Medicina de la Universidad Johns Hopkins (Baltimore, EE.UU.), Susan Wert y Jeffrey Whisett de la Unidad de Neonatología y Biología Pulmonar del Hospital Infantil Cincinnati Children's Hospital (Cincinnati, Ohio, EE.UU.) por el proceso de las muestras y estudio genético.
A la Dra. Ana Patiño (Laboratorio del Departamento de Pediatría de la Clínica Universitaria), que participó en el trabajo de laboratorio y en la discusión de los resultados.
