




Introducción
La epilepsia es una de las patologías neurológicas más comunes en la población pediátrica1-5. No obstante, a pesar de que la mayoría de los estudios epidemiológicos se han llevado a cabo en países industrializados y de condiciones socioeconómicas y sanitarias equivalentes; las cifras disponibles respecto a la incidencia anual de epilepsia infantil son muy variables, oscilando entre 41 y 100 casos por 100.0001,6-11, generalmente debido a razones metodológicas que hacen difícil la comparación de resultados. En España apenas existen datos relativos a la prevalencia y/o incidencia de la epilepsia infantil12,13.
La difusión de estudios epidemiológicos debería despertar gran interés en las administraciones públicas ya que el conocimiento de la incidencia de cualquier enfermedad, y en este caso de la epilepsia infantil, permitiría, por ejemplo, calcular y/o programar necesidades asistenciales de la población. La International League Against Epilepsy (ILAE), consciente de la conveniencia de disponer de referencias metodológicas comunes, ha propuesto una serie de pautas y/o normas conceptuales que hay que seguir en los estudios epidemiológicos14,15.
El objetivo del presente trabajo consiste en calcular la incidencia y la distribución relativa de las distintas epilepsias y síndromes epilépticos infantiles en nuestro medio, aplicando las recomendaciones de la ILAE.
Pacientes y métodos
La Comunidad Foral de Navarra tiene una población total de 555.829 habitantes (INE, censo de población, 2001), con una población infantil (menores de 15 años) de 76.236 (varones: 39.252 [51,5 %]; mujeres: 36.984 [48,5 %]). El Hospital Materno-Infantil Virgen del Camino de Pamplona (HVC) es el centro de referencia de patología neuropediátrica de Navarra, donde están ubicadas las Unidades de Neuropediatría y Neurofisiología. La organización estructural y/o funcional del Sistema Navarro de Salud facilita que todos aquellos pacientes con sospecha de padecer crisis convulsivas y/o epilepsia sean remitidos sin demora desde los Centros de Salud u hospitales secundarios, ubicados en las poblaciones de Tudela y Estella, al hospital de referencia donde se realiza una valoración neuropediátrica y un seguimiento evolutivo y, en consecuencia, el diagnóstico sindrómico de la práctica totalidad de pacientes pediátricos con epilepsia de nuestra comunidad.
De manera prospectiva se han registrado todos los pacientes menores de 15 años diagnosticados de epilepsia y residentes en Navarra en el período de tiempo comprendido entre enero de 2002 y diciembre de 2005 (4 años). De cada paciente se obtuvieron datos epidemiológicos (sexo, edad al diagnóstico, antecedentes personales y familiares de convulsiones febriles y epilepsia) y datos clínicos (tipo de crisis y hallazgos neurológicos y/o patología asociada), junto a exámenes complementarios (electroencefalograma [EEG] y estudios de neuroimagen: tomografía computarizada [TC] y/o resonancia magnética [RM] craneal) y, en su caso, estudios genéticos, metabólicos y/o neurofisiológicos en orden a establecer la etiología idiopática, sintomática o criptogénica de la epilepsia.
Para el diagnóstico y clasificación de las epilepsias y síndromes epilépticos se han aplicado los criterios de la ILAE15,16. Fueron descartados aquellos pacientes que presentaron crisis convulsivas exclusivamente en el período neonatal. Los diagnósticos sindrómicos correspondientes a cada paciente fueron discutidos y consensuados por los componentes de la Unidad de Neuropediatría.
Los resultados se expresan como medias y porcentajes e intervalos de confianza (IC del 95 %). La tasa de incidencia anual se expresa por 100.000 habitantes del mismo grupo de edad. Para el análisis estadístico (chi cuadrado y comparación de proporciones) se ha utilizado el programa informático Sigma-Plus (Hardware, 97).
Resultados
Durante los 4 años del período de estudio (2002-2005) fueron diagnosticados 191 casos nuevos de epilepsia entre la población infantil residente en Navarra. La distribución anual de casos nuevos fue muy regular (año 2002: 49 casos, año 2003: 46 casos; año 2004: 47 casos, y año 2005: 49 casos). La distribución de los pacientes por grupos de edad fue de 22 lactantes (de 1 mes hasta 12 meses), 66 preescolares (de 1 hasta 6 años), 54 escolares (de 6 hasta 10 años) y 49 adolescentes (de 10 hasta 15 años).
La etiología de la epilepsia fue considerada como idiopática en 78 casos (40,8 %), criptogénica en 62 (32,5 %) y sintomática en 51 (26,7 %). En los lactantes, la mayoría de las epilepsias eran sintomáticas (63,6 %) y, en menor proporción, criptogénicas (27,3 %) e idiopáticas (9,1 %). En los preescolares, la mayoría eran criptogénicas (43,9 %) e idiopáticas (30,3 %) y, en menor proporción, sintomáticas (25,8 %). En los escolares, la mayoría eran idiopáticas (57,4 %) y, en menor proporción, criptogénicas (27,8 %) y sintomáticas (14,8 %). Y en los adolescentes, la mayoría eran idiopáticas (55,1 %) y, en menor proporción, criptogénicas (26,5 %) y sintomáticas (18,4 %).
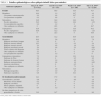
En la tabla 1 se exponen y comparan las características epidemiológicas en relación con los distintos grupos etiológicos. No existían diferencias significativas en la distribución por sexos en ninguno de los grupos etiológicos, siendo la relación varón:mujer de 1:2. Sin embargo, existían diferencias estadísticamente significativas (p < 0,05) en la distribución de los grupos etiológicos en relación con los grupos de edad; por ejemplo, los lactantes que representaban el 11,5 % del total de la muestra suponían el 3,8 % de las epilepsias idiopáticas y el 31,4 % de las sintomáticas. El 13,6 % de los pacientes referían antecedentes personales y/o familiares de convulsiones febriles y el 24 % de epilepsia familiar; pero ambos antecedentes se daban preferentemente y de manera estadísticamente significativa (p < 0,05) en las epilepsias idiopáticas y/o criptogénicas.
Se realizó TC craneal a 45 pacientes (23,6 %), RM craneal a 100 (52,3 %) y ambas pruebas a 37 pacientes (19,3 %). Es decir, se realizaron pruebas de neuroimagen en el 95,2 % de los pacientes (n = 182), y particularmente RM craneal al 71,7 % (n = 137). Los 9 pacientes en los que no se practicaron pruebas de neuroimagen (4,7 %) fueron diagnosticados de ausencias infantiles (2 casos), epilepsia rolándica (4 casos), epilepsia generalizada secundaria a síndrome específico: síndrome de Angelman (un caso) y enfermedad de Tay-Sachs (un caso), y epilepsia con crisis tónico-clónicas generalizadas del despertar (un caso).
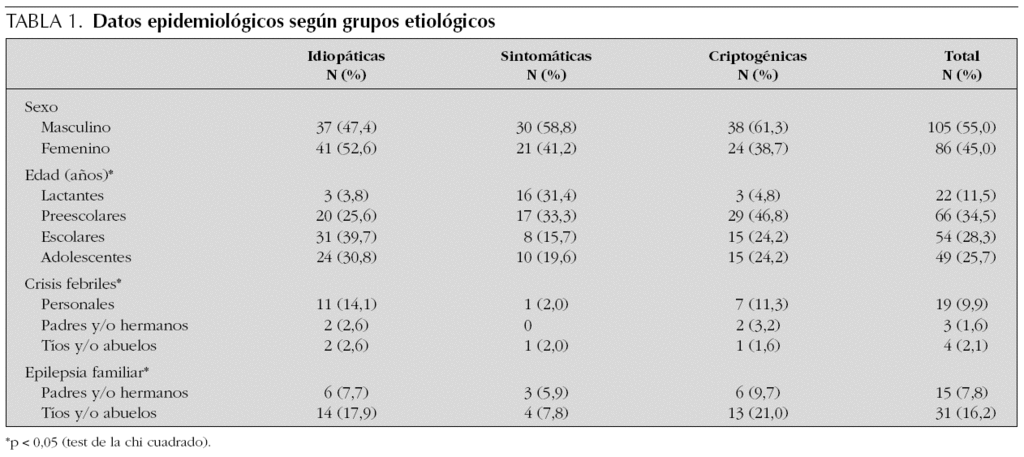
En la tabla 2 se exponen las tasas de incidencia anual de epilepsia por grupos de edad. La tasa de incidencia global era de 62,6 por 100.000/año (IC 95 %: 62,3-62,9). La tasa de incidencia en el primer año de vida fue de 95,3 (IC 95 %: 82,8-107,8), en los preescolares de 63,6 (IC 95 %: 61,4-65,8), en los escolares de 69,7 (IC 95 %: 61,1-78,3), y en los adolescentes de 48,7 (IC 95 %: 45,4-52,0). La incidencia era mayor en los varones respecto a las mujeres (66,8 frente a 58,1), pero sin que existieran diferencias estadísticamente significativas entre ambos sexos.
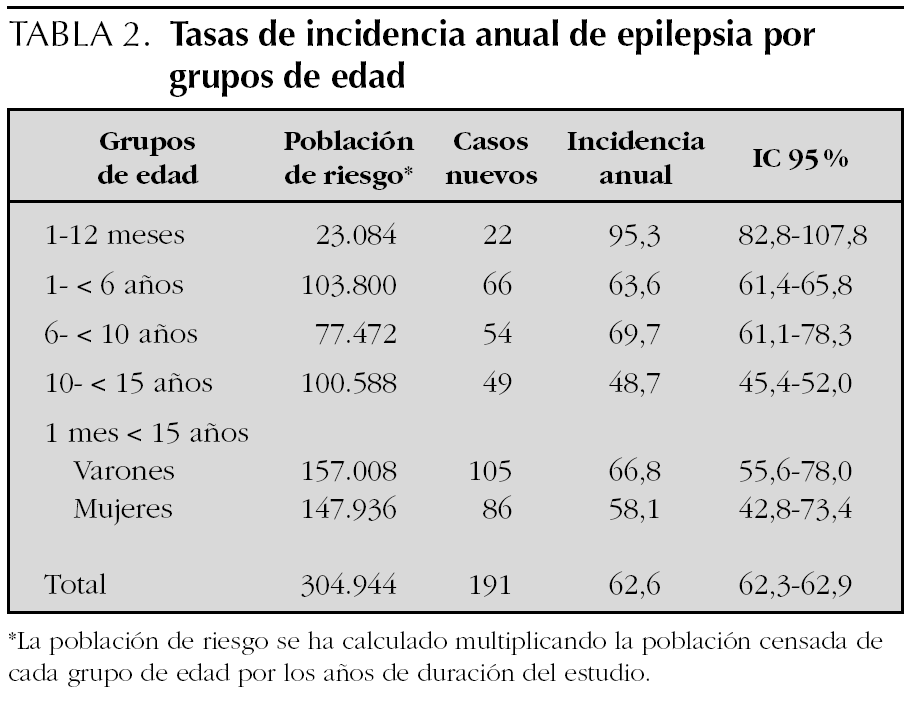
En la tabla 3 se expone la distribución de las distintas epilepsias y síndrome epilépticos en relación con la edad. El 55 % de los pacientes (n = 105) presentaban epilepsias focales, el 42,9 % (n = 82) generalizadas y en el 2,1 % restante (n = 4) eran de localización indeterminada. En los lactantes, el síndrome de West (45,5 %), las epilepsias asociadas a síndromes específicos (27,3 %) y las epilepsias focales sintomáticas (13,6 %) eran los síndromes epilépticos más prevalentes. En los preescolares, lo eran las epilepsias focales sintomáticas (22,7 %) o criptogénicas (21,2 %) y el síndrome de Doose (13,6 %). En los escolares, las epilepsias focales benignas (27,8 %) y criptogénicas (18,5 %) y las ausencias (18,5 %). Y en los adolescentes, las epilepsias focales criptogénicas (27,6 %) y benignas (18,4 %). Existía una relación inversa entre los grupos de edad y el tipo de crisis, a menor edad las epilepsias eran preferentemente generalizadas (81,8 % en el grupo de lactantes) y a mayor edad lo eran preferentemente focales (59,2 % en el grupo de escolares y de adolescentes).
Discusión
El tamaño de la población evaluada es un factor determinante de la variabilidad de las tasas de incidencia. La población infantil navarra podría considerarse como de tamaño apropiado para este tipo de estudios, ya que al no ser excesivamente grande y, por otra parte, gozar de un sistema sanitario muy estructurado, las posibilidades de obtener un registro completo de todos los pacientes son máximas y, desde luego, mínimas las dificultades para la aplicación de criterios uniformes en el diagnóstico y seguimiento de los casos; lo que, junto con la aplicación de las recomendaciones de la ILAE, acreditaría la validez de los resultados obtenidos.
La tasa de incidencia anual de epilepsia observada en la población infantil de Navarra (tabla 4) coincide con los datos aportados por diversos estudios metodológicamente comparables6,8,11. No obstante, existen autores que refieren menores tasas de incidencia, que oscilan entre 41 y 45/100.000, pero se trata de estudios retrospectivos y/o con grupos de edades distintos y presumiblemente con una recogida incompleta de casos9,10,13. En este trabajo, al igual que en la mayoría de estudios, la tasa de incidencia fue máxima durante el primer año de vida para luego ir disminuyendo gradualmente hasta alcanzar en la adolescencia las menores tasas de incidencia. La incidencia fue mayor en los varones, pero las diferencias entre ambos sexos no eran significativas.
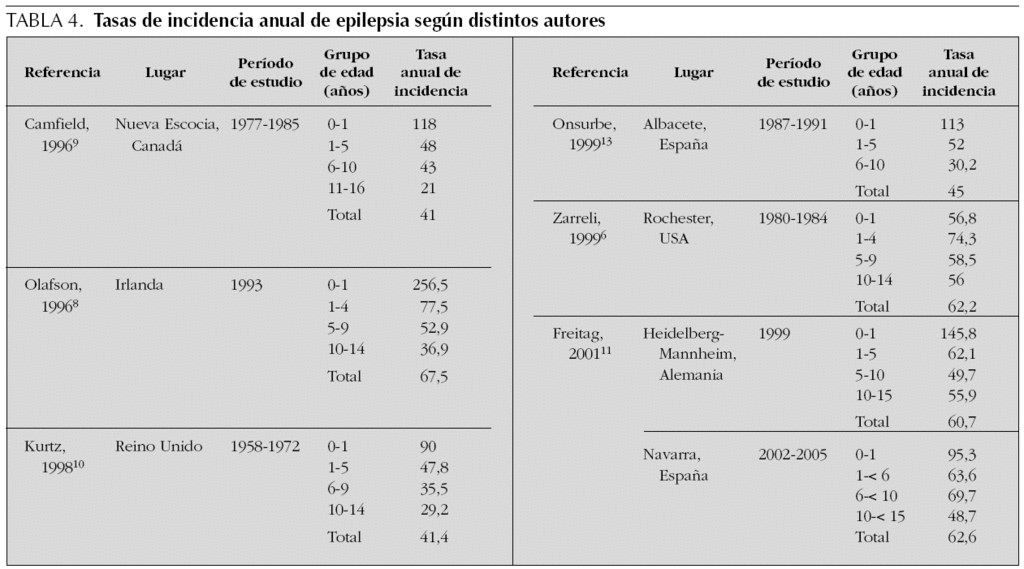
La edad representa un factor determinante en la fisiopatología de los distintos tipos de crisis y síndrome epilépticos, ya que las modificaciones estructurales y funcionales que el cerebro va experimentando desde el nacimiento hasta la adolescencia condicionan la expresión clínica y neurofisiológica de las epilepsias17,18. De hecho, tal y como ocurría en esta serie, la distribución relativa de los tipos de crisis y síndromes epilépticos era diferente en cada grupo de edad. Por ejemplo, en los primeros años de vida, especialmente en los lactantes, predominaban las epilepsias generalizadas; mientras que, a medida que los grupos eran de mayor edad predominaban las epilepsias focales. Además, algunos síndromes epilépticos se presentaban de forma predominante en determinados grupos de edad, tal como ocurría con el síndrome de West en los lactantes, el síndrome de Doose en los preescolares, las epilepsias focales benignas y ausencias en los escolares y las epilepsias focales criptogénicas en los adolescentes. Por tanto, al hablar de la epilepsia infantil sería preceptivo hacer referencia al grupo de edad de los pacientes que manejamos, puesto que las características epidemiológicas de las distintas epilepsias y síndromes epilépticos en la infancia guardan una estrecha relación con el nivel de maduración cerebral alcanzado.
A pesar de la aplicación de los criterios de la ILAE, los datos epidemiológicos publicados en los últimos años sobre epilepsia infantil son muy discordantes2,3,6,11,13,19-24. En la tabla 5 se exponen las frecuencias relativas de los distintos síndromes epilépticos referidos por distintos autores con evidentes diferencias, especialmente en lo relativo a las epilepsias catalogadas en categorías inespecíficas, tales como otras epilepsias no definidas previamente focales y/o generalizadas y las epilepsias de localización indeterminada sin datos inequívocos de crisis focales o generalizadas. En este estudio, apenas un 9,4 % de casos fueron catalogados como epilepsias en categorías inespecíficas: 12 casos (6,3 %) de epilepsias generalizadas idiopáticas, 2 casos (1 %) de epilepsias generalizadas sintomáticas y 4 casos (2,1 %) de localización indeterminada; mientras que otros autores refieren hasta un 8,6 % (24) y 14,7 % (6) de epilepsias de localización indeterminada sin datos inequívocos de crisis focales o generalizadas, así como elevadas proporciones de epilepsias sin clasificar y/o definir11,13,22,24. Estas diferencias podrían comprenderse al tratarse de epilepsias y/o síndromes epilépticos relativamente poco definidos y, por tanto, sujetos a diversas interpretaciones; sin embargo, también existen grandes discrepancias en relación otros síndromes epilépticos bien diferenciados, tales como las epilepsias focales benignas y/o las ausencias infantiles. Estas notorias diferencias advierten de la complejidad que entraña un diagnóstico sindrómico en la infancia, ya que la variabilidad de los hallazgos clínicos y electroencefalográficos que suelen presentar estos pacientes condicionan que, incluso entre observadores cualificados, resulte difícil establecer las características diferenciales de los distintos síndromes epilépticos, sobre todo al comienzo de la enfermedad25-29.
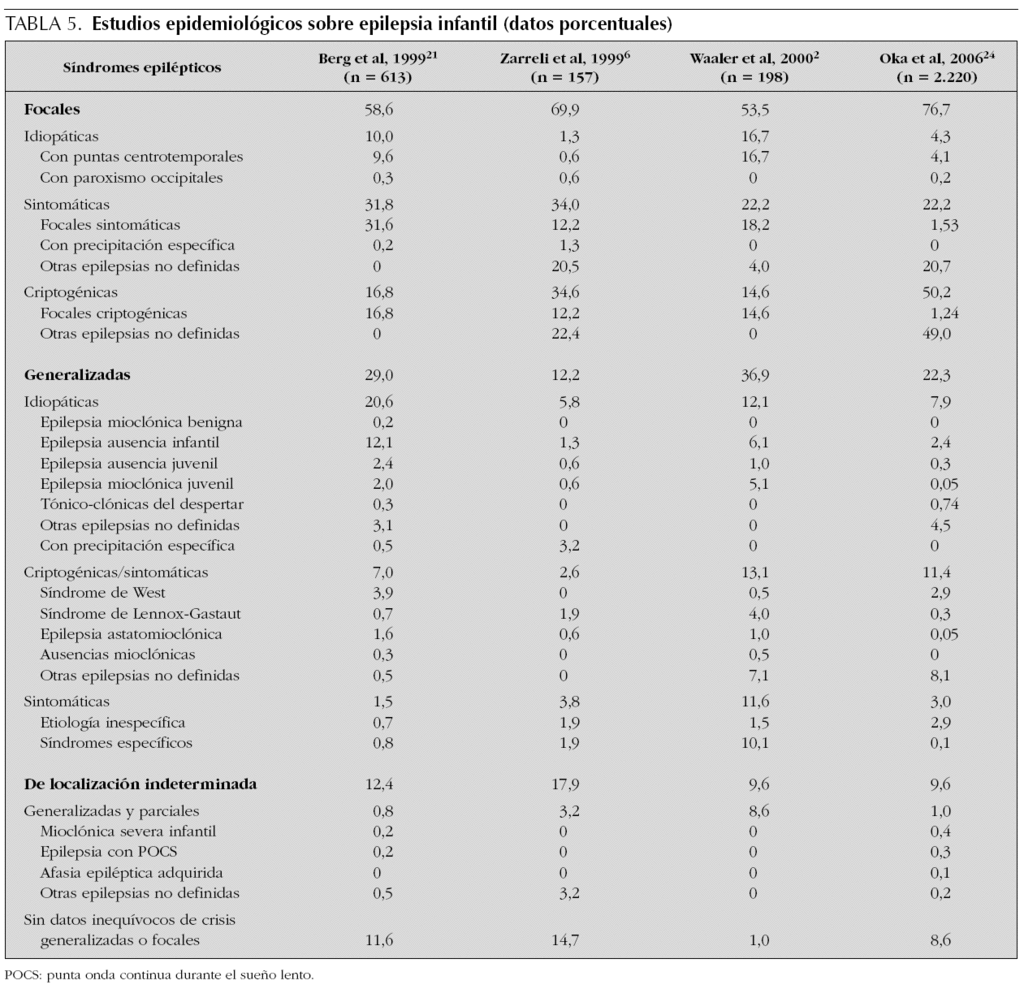
En resumen, la incidencia anual de la epilepsia infantil en Navarra (62,6 casos/100.000) es similar a la referida en diversos países occidentales; con una incidencia máxima en el primer año de vida (95,3 casos/100.000) que disminuye gradualmente hasta la adolescencia (48,7 casos/100.000). No obstante, sería conveniente diseñar estudios multicéntricos que confirmasen estos resultados en orden al cálculo racional de las necesidades asistenciales neuropediátricas de la población infantil. Además, cabe indicar que si bien en este estudio, el 55 % de los pacientes presentaban epilepsias focales, el 42,9 % generalizadas y el 2,1 % restante de localización indeterminada; los datos publicados respecto a la frecuencia relativa de las epilepsias y síndromes epilépticos son muy discordantes, lo que advierte de la dificultad intrínseca para establecer las características diferenciales de las distintas epilepsias y síndromes epilépticos y de la necesidad de disponer y aplicar criterios uniformes en orden a obtener datos epidemiológicos válidos y comparables.
Correspondencia: Dr. T. Durá Travé.
Unidad de Neuropediatría. Hospital Virgen del Camino.
Avda. Pío XII, 10, 8 °C. 31008 Pamplona. España.
Correo electrónico: tduratra@cfnavarra.es
Recibido en noviembre de 2006.
Aceptado para su publicación en marzo de 2007.
