






Desde que en 1942 Albright y colaboradores describieran por primera vez el pseudohipoparatiroidismo como la existencia de hipocalcemia e hiperfosfatemia asociadas a resistencia tisular a la hormona paratiroidea (PTH) en presencia de una función renal normal, se han realizado grandes avances en la caracterización clínica y genética de los pacientes afectos de esta enfermedad. De hecho, no solo se han identificado las alteraciones moleculares responsables, sino que se ha podido establecer que variantes en otros genes de la misma vía de señalización, PTH/PTHrP a través de la proteína Gsα, son la causa de enfermedades que comparten determinadas manifestaciones clínicas con el pseudohipoparatiroidismo.
En el ámbito pediátrico, los primeros síntomas o signos que deben hacernos pensar en alteraciones en esta vía son la presencia de osificaciones subcutáneas, la braquidactilia y/o la obesidad de inicio precoz, seguidas en el tiempo por la posible aparición de resistencia a la PTH. Esta sospecha clínica deberá ser confirmada mediante un diagnóstico molecular que permita el correcto seguimiento clínico coordinado y multidisciplinar. Entre los aspectos a tener en cuenta en la atención de estos pacientes se incluye la evaluación al diagnóstico y seguimiento de la eventual presencia de resistencia a la PTH y a la hormona tirotropa (TSH), deficiencia de hormona de crecimiento (GH), hipogonadismo, alteraciones esqueléticas, alteraciones de la salud dental, obesidad, resistencia a la acción de la insulina, intolerancia a la glucosa o diabetes mellitus tipo2 e hipertensión, así como osificaciones ectópicas (subcutáneas o con afectación de tejidos más profundos) y alteración del desarrollo neurocognitivo.
Since Albright and co-workers described pseudohypoparathyroidism in 1942 as the combined presence of hypocalcaemia and hyperphosphataemia associated with the existence of tissue resistance to parathyroid hormone (PTH) action upon normal renal function, great advances have been made in the clinical and genetic profile of patients affected by this condition. Furthermore, not only have genetic bases of pseudohypoparathyroidism been unravelled, but also variants in other genes involved in the PTH/PTHrP signalling pathway through Gsα, have been identified as the cause of diseases that share clinical features with pseudohypoparathyroidism.
In the paediatric setting, the first symptoms suggesting the impairment of this signalling pathway are the presence of subcutaneous ossifications, brachydactyly and/or early onset obesity, followed by the possible development of PTH resistance. This clinical suspicion should be confirmed by an accurate molecular diagnosis to allow for coordinated multidisciplinary clinical management. Among the features of this group of disorders, physicians should pay attention to evaluation of PTH and/or thyrotropin (TSH) resistance at diagnosis and throughout follow-up, as well as growth hormone deficiency, hypogonadism, skeletal deformities, dental impairment, obesity, insulin resistance, impaired glucose tolerance or type2 diabetes mellitus and hypertension, as well as ectopic ossifications (either subcutaneous or affecting deeper tissues) and impairment of neurocognitive development.
El pseudohipoparatiroidismo (PHP) y sus enfermedades relacionadas engloban a un grupo de patologías caracterizadas por hallazgos físicos que suelen incluir, con frecuencia variable, acortamientos óseos predominantemente acromélicos, talla baja, obesidad de inicio precoz y osificaciones ectópicas, así como alteraciones endocrinológicas que incluyen resistencia a la hormona paratiroidea (PTH) y estimulante del tiroides (TSH). Estas enfermedades están causadas por alteraciones genéticas y/o epigenéticas en la vía de señalización de PTH/PTHrP a través de la proteína Gsα1.
Este documento es una versión abreviada, dirigida a las consideraciones y recomendaciones más relevantes en la edad pediátrica, del primer consenso internacional para el diagnóstico y atención a pacientes con pseudohipoparatiroidismo y enfermedades relacionadas, recientemente publicado en la revista Nature Reviews Endocrinology2. Es reseñable la participación de representantes de las asociaciones de pacientes en la elaboración de este documento de consenso, entre ellas, de la asociación española (asociacionpacientesphp@gmail.com).
Diagnóstico clínicoEl PHP y sus enfermedades relacionadas presentan una considerable variabilidad clínica. Algunas de sus manifestaciones pueden pasar desapercibidas con frecuencia en niños muy pequeños, principalmente las osificaciones, la braquidactilia o la hipocalcemia, que suelen apreciarse con el paso del tiempo.
Definiciones clínicasEl término «osteodistrofia hereditaria de Albright» (OHA) se emplea para identificar un conjunto de manifestaciones clínicas originariamente descritas por F. Albright y que incluyen: cara redondeada, hábito pícnico con talla baja, braquidactilia (que puede aparecer tardíamente) y osificaciones ectópicas3. El retraso en el desarrollo intelectivo se incluyó posteriormente como una característica adicional (aunque principalmente aparece en los pacientes con PHP1A); asimismo, la obesidad, particularmente la de inicio precoz, así como la macrocefalia relativa para la altura pueden ser parte del fenotipo4-6.
El PHP1A se definió inicialmente como la asociación de resistencia a múltiples hormonas, incluyendo PTH y TSH, fenotipo OHA y actividad Gsα disminuida en ensayos in vitro7. La resistencia a TSH suele ser detectada precozmente, incluso en el cribado neonatal, mientras que la resistencia a PTH se suele desarrollar posteriormente8,9. De forma similar, la braquidactilia se desarrolla progresivamente, haciéndose evidente habitualmente antes de la pubertad10. Se han descrito otras características adicionales en pacientes afectos de PHP1A (reducción de la longitud neonatal, obesidad de inicio precoz, alteraciones cognitivas). Es decir, es posible que un paciente con una obesidad muy precoz o con hipotiroidismo neonatal presente PHP1A, cuyo perfil clínico/bioquímico clásico (resistencia a la PTH y/o braquidactilia) aún no sea manifiesto y esto implique un retraso en el diagnóstico2.
El PHP1C se ha definido como la asociación de todas las características del PHP1A, pero con demostración de actividad normal Gsα en los ensayos de complementación in vitro11. Debido al solapamiento con el PHP1A y a la falta de estudio de la actividad Gsα en la mayor parte de publicaciones —y consecuentemente a la falta de diferenciación entre PHP1A y PHP1C— tanto en este documento como en el del consenso original, no se ha diferenciado específicamente el PHP1C, considerándolo una variante del PHP1A, tanto en el texto como en las recomendaciones, a menos que se señale lo contrario.
El PHP1B se describió inicialmente como la resistencia aislada a PTH, en ausencia de fenotipo OHA y con niveles normales de actividad Gsα. Algunos pacientes con PHP1B presentan características bioquímicas y algunos rasgos del fenotipo OHA superponibles al PHP1A (siendo difícil su diferenciación)12,13.
El pseudo-PHP (PPHP) se definió como la existencia de fenotipo OHA, con actividad Gsα disminuida, en ausencia de resistencia a PTH14. La presencia de osificaciones subcutáneas es frecuente (en forma de osteoma cutis o placas óseas) en estos pacientes15, y en algunos casos puede existir resistencia leve a PTH y TSH16.
La heteroplasia ósea progresiva (HOP) consiste en la presencia de osificaciones ectópicas que se extienden progresivamente en profundidad en el tejido conectivo. Los pacientes afectos de HOP habitualmente no manifiestan otros signos de OHA y presentan una sensibilidad normal a PTH. Las osificaciones pueden determinar graves anquilosis de las articulaciones implicadas e hipocrecimiento local17.
La acrodisostosis se define como la asociación de braquidactilia grave, disostosis facial e hipoplasia de la raíz nasal. La braquidactilia habitualmente afecta a las falanges, metacarpos y metatarsos, con la excepción de los primeros dedos de las extremidades. En las radiografías, las epífisis adoptan una apariencia cónica, la maduración esquelética está acelerada y estas alteraciones pueden estar ya presentes al nacimiento o poco tiempo después18.
El PHP tipo 2 se caracteriza por el incremento de AMPc en respuesta a la administración exógena de PTH pero con respuesta fosfatúrica deficiente19. La causa molecular de esta variante de la enfermedad es desconocida en el momento actual.
Principales características clínicas del pseudohipoparatiroidismo y enfermedades relacionadasEl diagnóstico de estas enfermedades en la etapa pediátrica puede ser difícil debido a que muchas de las manifestaciones que se asocian con ellas son poco específicas. El diagnóstico diferencial de los pacientes con obesidad, hipotiroidismo de inicio precoz (incluso congénito), talla baja y/o braquidactilia es muy amplio e incluye múltiples enfermedades endocrinológicas y entidades sindrómicas (tabla 1)2.
Diagnóstico diferencial del pseudohipoparatiroidismo (PHP) y enfermedades relacionadas basado en su característica clínica principal
| Síntoma principal | Diagnóstico diferencial | Signos asociados y comentarios |
|---|---|---|
| Hipocalcemia con elevación de PTH | Deficiencia de vitaminaD o resistencia a la misma | Mejoría tras tratamiento con vitaminaD. Raquitismo, alopecia |
| Raquitismo hipofosfatémico | Ensanchamiento metafisario. Genu valgum. Fosfatasa alcalina elevada e hipofosfatemia | |
| Hipoparatiroidismo debido a mutación en el gen PTH | Empleo de diferentes ensayos para confirmar la existencia de niveles séricos elevados de PTH | |
| Braquidactilia | Síndrome trico-rino-falángico debido a mutaciones en el gen TRPS1 | Estigmas: cabello escaso de crecimiento lento, colas de las cejas pequeñas, nariz bulbosa, filtrum largo y plano, labio superior fino y pabellones auriculares prominentes. Displasia de caderas, pies pequeños y primer metatarsiano corto, exostosis y epífisis «marfiladas» |
| Braquidactilia aislada tipoE asociada a mutaciones en el gen HOXD13 | Sindactilia, alargamiento de las falanges distales, con acortamiento de la del pulgar. Hipoplasia/aplasia, duplicación falángica lateral y/o clinodactilia. | |
| Síndrome de braquidactilia y retraso mental debido a microdeleciones en 2q37 | Obesidad, talla baja. Braquidactilia. Alteraciones cognitivas y del desarrollo psicomotor | |
| Síndrome de Turner (ausencia parcial o total de uno de los cromosomas X) | Talla baja, peso neonatal bajo, disgenesia gonadal con fallo ovárico, alteraciones neurocognitivas variables. Braquidactilia y deformidad de Madelung | |
| Braquidactilia tipoE con talla baja debido a mutaciones en PTHLH | Talla baja de intensidad variable, alteración en el desarrollo mamario. Oligodontia y retraso en la erupción dental, malposición dental. Pseudoepífisis. Braquidactilia | |
| Osificaciones subcutáneas | Acné vulgaris. | |
| Tumores cutáneos como: pilomatrixomas, siringomas condroides, carcinomas de células basales, quistes pilares y nevi. Osteoma cutis secundario o traumático y osteoma miliaria | ||
| Condiciones inflamatorias tales como cicatrices, estasis venosa crónica, morfea, esclerodermia, dermatomiositis o miositis osificante progresiva | ||
| Osificaciones progresivas | Fibrodisplasia osificante progresiva debido a mutaciones tipo missense activadoras recurrentes en ACVR1 | Osificación progresiva de músculos esqueléticos, tendones, fascias y ligamentos. La región dorsal alta y el cuello son las primeras regiones afectadas; los traumatismos modifican la progresión natural de la enfermedad. Malformaciones congénitas de los primeros dedos de los pies |
| Calcinosis tumoral debida a mutaciones en FGF23 o GALNT3 | Deposición de calcio en la piel o los músculos. Hiperfosfatemia | |
| Obesidad de inicio precoz | Síndrome de Beckwith-Wiedemann | Hemihipertrofia. Macroglosia |
| Anomalías genéticas, citogenéticas o sindrómicas asociadas a obesidad de inicio precoz tales como el síndrome de Prader-Willi. Obesidades de etiología monogénica (vía de señalización leptina-POMC, genes implicados en el desarrollo hipotalámico) | ||
| Hipotiroidismo de inicio precoz | Hipotiroidismo congénito de cualquier causa | Tiroides ortotópico de pequeño tamaño. Elevación moderada de TSH |
| Resistencia a TSH debida a mutaciones en el receptor de TSH | ||
| Hipertensión arterial | Síndrome autosómico dominante de hipertensión y braquidactilia tipoE | Talla baja |
PHP: pseudohipoparatiroidismo; POMC: proopiomelanocortina; PTH: hormona paratiroidea; TSH: hormona estimulante del tiroides.
La lista de diagnósticos diferenciales dista de ser exhaustiva, pero contempla las principales entidades que presentan solapamiento fenotípico con el PHP y sus enfermedades relacionadas.
En la mayor parte de pacientes con PHP las manifestaciones clínicas más significativas son los síntomas de hipocalcemia derivados de la resistencia a PTH20 (que suele ser de aparición tardía en la infancia)21. Los periodos de crecimiento acelerado (que determinan requerimientos incrementados de calcio) o la deficiencia nutricional de calcio o vitaminaD pueden desencadenar o intensificar los síntomas22.
Las primeras alteraciones bioquímicas en observarse suelen ser la elevación de la PTH y de la fosfatemia, seguidas por la hipocalcemia (con hipocalciuria y niveles normales o bajos de calcitriol), en una instauración progresiva a lo largo de meses, e incluso años23,24.
El hiperparatiroidismo secundario prolongado, con hipocalcemia crónica y deficiencia 1,25(OH)2 vitaminaD, se ha asociado con el desarrollo de hiperparatiroidismo terciario en pacientes adultos y con PHP1B25 (aunque pudiera observarse también en adolescentes) y puede ocasionar también la resorción ósea y desmineralización similares a los observados en el raquitismo.
La resistencia a PTH también está presente en pacientes con acrodisostosis causada por mutaciones en PRKAR1A, aunque la presencia de hipocalcemia no ha sido documentada. Los pacientes con mutaciones en PDE4D habitualmente presentan niveles normales de PTH26.
La osificación ectópica aparece en enfermedades causadas por la alteración molecular del gen GNAS tales como PHP1A/1C, PPHP y HOP. La osificación ectópica no es sinónimo de calcificación, ni se relaciona con la calcemia o la fosforemia. Puede ser detectada en forma de placas (osteoma cutis) al nacimiento o en los primeros años de vida de los pacientes, o como nódulos aislados en niños. Los nódulos pueden ser múltiples, duros a la palpación y de tamaño variable27,28. No existe evidencia de que las osificaciones sean consecuencia de traumatismos locales. Las osificaciones ectópicas se encuentran en el 100%, el 80-100%, el 30-60% y excepcionalmente en los pacientes afectos de HOP, PPHP/OHA, PHP1A y PHP1B, respectivamente23. Hasta la fecha no se ha comunicado presencia de osificaciones en los pacientes afectos de acrodisostosis. Las osificaciones que se desarrollan en la HOP son progresivas y se extienden a los músculos, tendones y ligamentos. Pueden estar circunscritas o ser más prominentes en un hemicuerpo17.
La braquidactilia en el PHP y enfermedades relacionadas puede ser clasificada como tipoE, definida como un acortamiento variable de los metacarpianos (sobre todo el 5.o, el 4.o y el 3.o) junto con, habitualmente, longitud normal de las falanges y ocasionalmente acompañada por metatarsianos relativamente cortos. La braquidactilia se desarrolla con el tiempo y puede no estar presente en etapas precoces de la vida10,29, excepto en pacientes con acrodisostosis18, y su frecuencia y gravedad varían entre las distintas enfermedades (siendo del 100% en pacientes con acrodisostosis). En cualquier caso, la braquidactilia no es específica del PHP y enfermedades relacionadas, y se puede encontrar en pacientes afectos de múltiples enfermedades30 (tabla 1).
Los pacientes con PHP1A frecuentemente (si no siempre) presentan niveles séricos elevados de TSH y niveles séricos de hormonas tiroideas normales o discretamente reducidos. Algunos pacientes presentan un hipotiroidismo franco. La elevación de TSH debido a resistencia a la misma puede estar presente al nacimiento y ser detectada en el cribado neonatal. En el PHP1B los niveles de TSH están en el límite alto de la normalidad o levemente elevados en el 30 al 100% de pacientes2,23,31. La resistencia a TSH puede observarse en los pacientes con acrodisostosis debida a mutaciones en PRKAR1A, pero no en aquellos con mutaciones en PDE4D26,32. Se han hallado niveles séricos elevados de calcitonina en una gran serie de pacientes afectos de PHP1A y PHP1B, mientras que la resistencia a las gonadotropinas parece mucho más sutil que la resistencia a otras hormonas (como TSH o PTH), considerándose que existe exclusivamente una insensibilidad parcial a las gonadotropinas2.
Además, existen otros signos que, por no determinar cambios hormonales cuantificables, algunas veces no son reconocidos como parte del espectro del PHP y de sus enfermedades relacionadas. De hecho, aunque estos primeros síntomas pueden estar ya presentes en la infancia temprana, el diagnóstico con frecuencia se demora hasta la pubertad en pacientes afectos de PHP1A o acrodisostosis, e incluso hasta la vida adulta en pacientes con PHP1B, a menos que la historia familiar sea positiva. La presencia de osificaciones puede acelerar el proceso diagnóstico en el caso de HOP/osteoma cutis. Entre estos signos son particularmente reseñables la afectación de la antropometría neonatal y la obesidad. La presencia de antropometría pequeña para la edad gestacional se ha descrito en pacientes con mutación en el alelo paterno de GNAS33 y en la mayoría de los pacientes con acrodisostosis y mutación en PRKAR1A o PDE4D26,32. La mayoría de estos pacientes alcanzan una talla adulta baja (la talla media en PPHP u OHA es inferior a −2SDS), mientras que los pacientes con cambios en el patrón de metilación del locus GNAS muestran un moderado incremento del peso y de la longitud neonatales, y aunque existen escasas comunicaciones referentes a su talla adulta, esta parece situarse en el rango normal34.
Aunque sea asimismo una característica inespecífica, el desarrollo de obesidad se asocia con enfermedades relacionadas con el PHP, tales como el PHP1A/1C o la acrodisostosis. Es reseñable que la obesidad en estos pacientes es menos llamativa en la etapa adulta que en la infancia. Los pacientes con PHP1A pueden mostrar también progresivamente alteración en su sensibilidad a la acción de la insulina5,35,36.
Recomendamos estar atentos a estas manifestaciones, dado que un diagnóstico preciso puede conllevar implicaciones extremadamente importantes para una adecuada atención, ya que permite: a)el cribado y tratamiento precoz de las eventuales complicaciones endocrinológicas asociadas, tales como la resistencia a TSH o PTH; b)la prevención y el tratamiento de la obesidad o la talla baja; c)el adecuado tratamiento de las osificaciones ectópicas, y d)un adecuado consejo genético y prenatal.
Evolución de la clasificación de las enfermedades relacionadas con el pseudohipoparatiroidismoEl sistema de clasificación actual distingue variantes del PHP tales como PHP1A, PHP1B, PHP1C y PPHP. Como se ha mencionado, la asignación de los subtipos clásicos se basa en la presencia de fenotipo OHA junto con la caracterización de las resistencias hormonales y la determinación en ensayos in vitro de la actividad proteica de Gsα. Sin embargo, esta clasificación excluye entidades como la acrodisostosis o la HOP, cuyos fenotipos clínicos a veces se solapan. Este solapamiento hace que el diagnóstico sea complejo y la clasificación, de algún modo, no adaptada al conocimiento actual. Además, las herramientas disponibles para el estudio de los pacientes han evolucionado, siendo difícil el acceso a la determinación de la actividad Gsα en membrana celular, y convirtiéndose el diagnóstico molecular en la herramienta diagnóstica de referencia para la diferenciación de las variantes de PHP. Si bien hay una reciente propuesta de nueva clasificación para estas entidades clínicas1, es necesaria la validación de la misma.
Diagnóstico molecularLa figura 1 expone, en forma de algoritmo secuencial, las recomendaciones para el estudio y diagnóstico molecular del PHP y enfermedades relacionadas.
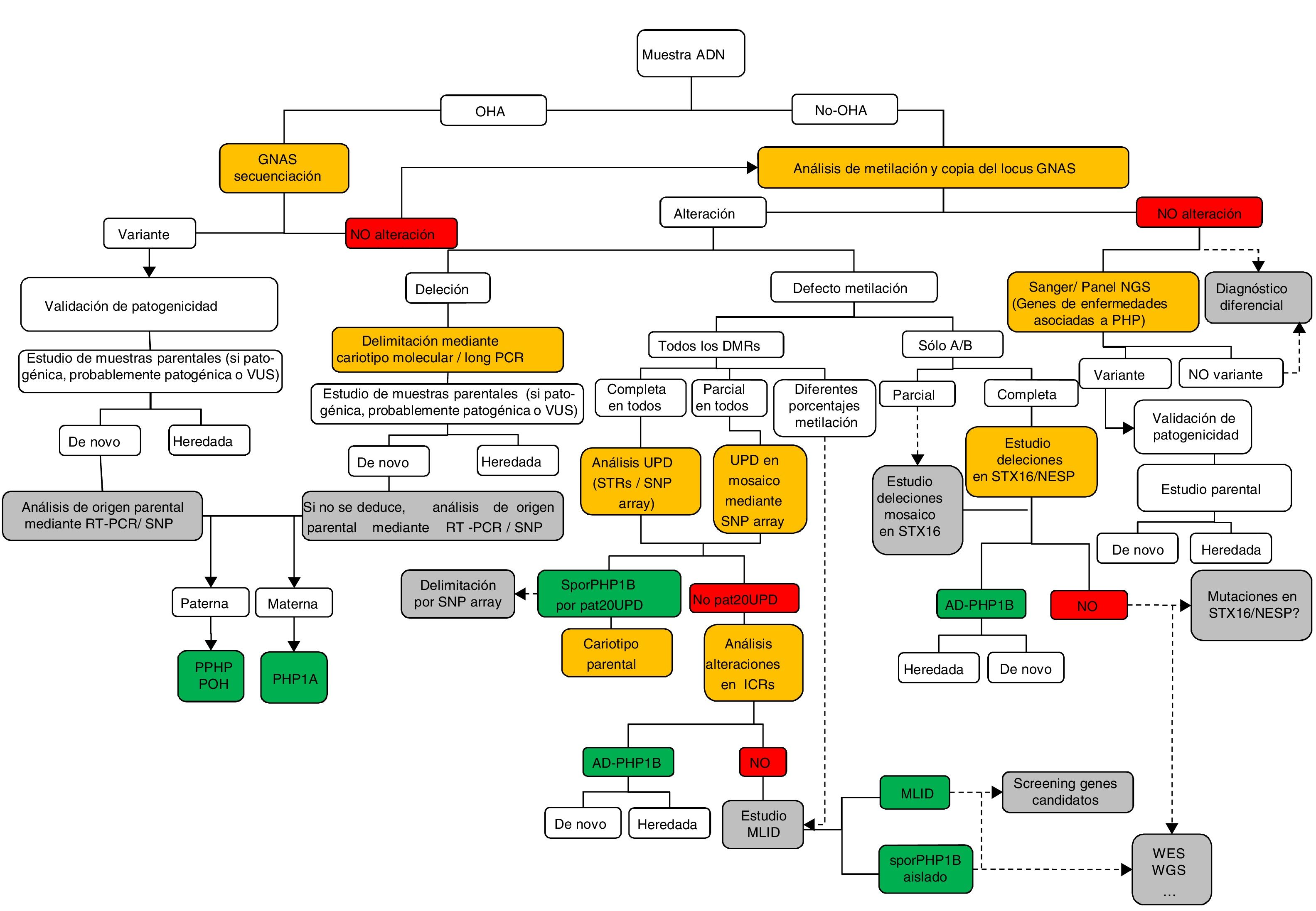
Algoritmo del estudio molecular para la confirmación del diagnóstico de PHP y enfermedades relacionadas.
Si el paciente presenta osteodistrofia hereditaria de Albright (OHA), hay que descartar la presencia de alteraciones genéticas en GNAS, incluyendo mutaciones puntuales (secuenciación) y reordenamientos genómicos. Una vez que la variante ha sido identificada, debe confirmarse su patogenicidad de acuerdo con las guías clínicas40 y, cuando sea posible, debe determinarse el origen parental.
En ausencia de OHA, el estudio debe comenzar por la búsqueda de alteraciones epigenéticas. En función del patrón de metilación obtenido, se necesitarán estudios posteriores para alcanzar el diagnóstico definitivo: a)si el defecto de la metilación está restringido a GNAS A/B:TSS DMR, deberán buscarse las deleciones de STX16 y, si se encuentran, se confirmará el diagnóstico de PHP1B autosómico dominante (AD-PHP1B); b)si la metilación afecta a los cuatro DMR, debería analizarse la posibilidad de disomía uniparental paterna del cromosoma 20 (patUPD20q); c)si no se encontrara la patUPD20q, deberían estudiarse las deleciones en NESP/AS; d)si no se identifican causas genéticas subyacentes a las alteraciones de la metilación, es probable que el paciente presente la forma esporádica de PHP1B (spor-PHP1B).
Una vez descartadas las alteraciones en el locus GNAS como la causa del fenotipo, y en pacientes con OHA, deberían secuenciarse los genes responsables de las enfermedades relacionadas a PHP (es decir, al menos PDE4D y PRKAR1A).
Los cuadros en amarillo indican la tecnología; en verde, la confirmación molecular definitiva; en gris se sugieren los pasos de investigación/futuros a realizar.
MLID: trastornos causados por alteración de la impronta en múltiples loci (multi-locus imprinting disturbance); NGS: secuenciación masiva (next-generation sequencing); STRs: microsatélites (short tandem repeats); UPD: disomía uniparental; VUS: variante de significado incierto; WES: secuenciación de exoma (whole exome sequencing); WGS: secuenciación de genoma completo (whole genome sequencing).
En pacientes con sospecha clínica de PHP se recomienda que el estudio molecular incluya el análisis de la secuencia, el estado de metilación y la presencia de alteraciones en el número de copias del locus GNAS2.
En el ámbito pediátrico, lo más común es que los pacientes se diagnostiquen por manifestar alguna(s) de las características del fenotipo de OHA (principalmente talla baja y/u obesidad de inicio precoz), por lo que se debería empezar el análisis por la secuenciación del gen GNAS21. Se debe proceder de forma similar en caso de que se detecte la presencia de osificaciones ectópicas2. En el caso de haberse identificado la variante causal, es importante determinar si esta se localiza en el alelo paterno o materno de cara a un correcto seguimiento clínico. Para ello, se realizará estudio de los progenitores o, en el caso de ser una variante de novo, caracterización mediante RT-PCR o SNP.
Cuando el paciente no presenta OHA, por lo general será diagnosticado debido a los síntomas causados por la hipocalcemia21, y deberá iniciarse el estudio molecular por la determinación del estado de metilación del locus GNAS. Si se detectara un patrón de metilación alterado, deben analizarse las posibles causas genéticas subyacentes (deleciones en STX16, NESP/AS o disomía uniparental) para un correcto asesoramiento genético.
En casos de braquidactilia severa, con afectación de todos los huesos de la mano, observada en los primeros años de vida del paciente, se ha de descartar la posibilidad de acrodisostosis mediante el análisis de la secuencia de los genes PRKAR1A y PDE4D26.
Atención y seguimientoActualmente no existen ensayos clínicos prospectivos centrados en el seguimiento y el tratamiento de los pacientes con PHP y enfermedades relacionadas. Las propuestas de intervención que se muestran en la tabla 2 son el fruto de la revisión de la bibliografía disponible hasta el desarrollo del consenso y de la experiencia de los participantes. Si bien en la tabla 3 se pueden encontrar todas las recomendaciones correspondientes a este apartado, resumiremos en las próximas líneas los aspectos más significativos para los pacientes pediátricos.
Resumen de las principales intervenciones a realizar en el ámbito pediátrico durante el seguimiento de los pacientes con pseudohipoparatiroidismo y enfermedades relacionadas
| Nacimiento a 2años | Infancia temprana, 2-6 años | Infancia tardía-adolescencia | |
|---|---|---|---|
| Guía anticipatoria | |||
| Refuerzo familiar | • | • | • |
| Asesoramiento genético | Al diagnóstico | Al diagnóstico | Al diagnóstico |
| Evaluación médica | |||
| Crecimiento lineal | • | • | • |
| Ganancia de peso / IMC | • | • | • |
| Descenso testicular | • | • | Si no se ha estudiado antes |
| Presión arterial | N/A | •* | • |
| Desarrollo/cognición | • | • | S |
| Evaluación psicosocial | N/A | • | S |
| Osificaciones ectópicas | • | • | |
| Ortodóntico/dental | N/A | • | • |
| Radiografía edad ósea | N/A | • (en caso de deceleración del crecimiento) | • (en caso de deceleración del crecimiento) |
| Metabolismo fosfocálcico | • | • | • |
| Tiroides | • | • | • |
| Pubertad | N/A | N/A | (bioquímica en caso de retraso) |
| Eje GH / IGF | N/A | • | • |
| Metabolismo hidrocarbonado y lipídico | N/A | • | • |
| Fertilidad | N/A | N/A | S |
• A realizar al diagnóstico y, posteriormente, anualmente; N/A: no aplicable; S: subjetivo (según historia clínica y examen físico).
Recomendaciones para la atención y cuidados de pacientes con pseudohipoparatiroidismo y enfermedades relacionadas
| Atención y cuidados |
|---|
| 3.1. El diagnóstico y la atención a los pacientes con PHP y enfermedades relacionadas deben ser realizados por un equipo multidisciplinar de especialistas |
| Manejo de la resistencia a PTH |
|---|
| 3.2. Al diagnóstico, o antes de iniciar el tratamiento, recomendamos monitorizar los niveles séricos de PTH, calcio, fósforo y 25-OH-vitaminaD. La medición de PTH, calcio y fósforo debe ser realizada regularmente (cada 6meses en niños y al menos anualmente en adultos) con la excepción de pacientes portadores de mutación bien en el alelo paterno de GNAS o en PDE4D, en los cuales, aparte de al diagnóstico, no es necesaria evaluación rutinaria posterior. La monitorización de los niveles de calcio debe ser más frecuente en individuos sintomáticos, durante las fases de crecimiento acelerado, enfermedad aguda y durante el embarazo y la lactancia, cuando las dosis necesarias de metabolitos activos de la vitaminaD o análogos pueden cambiar. Los niveles de 25-OH-vitaminaD deben normalizarse y mantenerse en el rango normal en todos los pacientes 3.3. La hipocalcemia severa sintomática requiere corrección inmediata, que puede incluir administración intravenosa de sales de calcio, de acuerdo con las guías generales de tratamiento de la hipocalcemia aguda en el hipoparatiroidismo. Estos pacientes deben ser tratados de forma concomitante con metabolitos activos de la vitaminaD (calcitriol) o análogos (alfa-calcidol) 3.4. En pacientes con elevaciones sustanciales y progresivas de los niveles de PTH e hiperfosfatemia se puede considerar el tratamiento con metabolitos activos de la vitaminaD o análogos, independientemente de la presencia de hipocalcemia. Los suplementos de calcio deberían plantearse en función de la ingesta diaria de calcio. Los niveles séricos de fósforo deben ser vigilados durante el tratamiento con metabolitos de la vitaminaD o análogos y suplementos de calcio 3.4.b. El tratamiento con metabolitos activos de la vitaminaD o análogos se puede plantear también cuando los niveles de PTH están por encima del doble del rango superior de la normalidad, independientemente de la presencia de hipocalcemia 3.5. Los objetivos del manejo convencional de la resistencia a la PTH incluyen el mantenimiento de unos niveles de calcio y fósforo dentro del rango normal, mientras se evita la hipercalciuria (corregida por tamaño y edad) y se disminuyen los niveles de PTH tanto como lo permitan los niveles de calcio en sangre y orina. Recomendamos el uso del metabolito activo de la vitaminaD calcitriol o el análogo activo de la vitaminaD (alfa-calcidol) con o sin suplementos de calcio, como el eje del tratamiento de la hipocalcemia crónica. Los pacientes no deberían ser tratados con PTH o con análogos de PTH. Durante el tratamiento, los niveles de PTH, calcio y fósforo deberían ser monitorizados cada 6meses en pacientes asintomáticos y más frecuentemente cuando esté clínicamente indicado. Los pacientes y/o sus familias deben ser instruidos acerca de los síntomas y signos de la hipo- y la hipercalcemia 3.6. Recomendamos estudios de imagen apropiados para evaluar la presencia de nefrocalcinosis durante la transición a la etapa adulta 3.7. Recomendamos estudios de imagen adecuados para la edad, para monitorizar el desarrollo o empeoramiento de la nefrocalcinosis en pacientes con hipercalciuria persistente en mediciones repetidas y cuando esté clínicamente indicado 3.8. Para la evaluación de las consecuencias a largo plazo de la hipocalcemia y la hiperfosfatemia, la TAC cerebral está indicada únicamente en presencia de manifestaciones neurológicas. El examen oftalmológico se recomienda para diagnosticar o excluir la presencia de cataratas o calcificaciones retinianas 3.9. Los quelantes del fósforo (diferentes del calcio) están muy raramente (si alguna vez lo están) indicados en el tratamiento de la hiperfosfatemia severa y persistente a largo plazo 3.10. Recomendamos revisiones dentales regulares cada 6-12meses durante la infancia y según lo clínicamente indicado en adultos |
| Manejo de la resistencia a TSH |
|---|
| 3.11. Se recomienda la evaluación al diagnóstico de la función tiroidea (incluyendo los autoanticuerpos en adolescentes y adultos) para la detección precoz de la resistencia a TSH e intervención precoz en todos los pacientes con PHP y enfermedades relacionadas. A partir de entonces, se recomienda la monitorización de TSH cada 6meses en pacientes menores de 5años de edad y anual en niños mayores y adultos 3.12. La indicación o indicaciones de tratamiento del hipotiroidismo, dosificación de levotiroxina y objetivos terapéuticos deben ser los mismos que para cualquier paciente con hipotiroidismo o hipotiroidismo subclínico |
| Manejo de las alteraciones del crecimiento y deficiencia de GH |
|---|
| 3.13. Recomendamos monitorización estrecha de la talla en niños en todos los exámenes de control (al menos cada 6meses) hasta que se alcance la talla final 3.14. Recomendamos la monitorización de la maduración esquelética utilizando radiografía simple en todos los niños (a excepción de aquellos con PHP1B) y evaluación de deficiencia de GH en todos los niños en el contexto de desaceleración del crecimiento longitudinal. Dado que la mayoría de los pacientes que desarrollan la deficiencia de GH lo hacen debido a resistencia a GHRH, la evaluación clínica y/o bioquímica del eje GH-IGF-1 debe ser realizada en todos los pacientes, típicamente alrededor de los 3-6años de edad y repetirse posteriormente según sea necesario 3.15. Los pacientes con deficiencia de GH deben ser tratados con rhGH. Son aún necesarios datos relativos a los resultados del tratamiento con GH en niños sin deficiencia de GH antes de poder recomendar este tratamiento también en estos pacientes 3.16. El tratamiento con rhGH podría plantearse para pacientes adultos con deficiencia de GH; sin embargo, no existe confirmación del beneficio en esta población y el tratamiento debe ser dado de acuerdo con las regulaciones específicas de cada país 3.17. Los pacientes con PHP que han nacido pequeños para la edad gestacional y que no muestran un crecimiento recuperador adecuado podrían ser candidatos a tratamiento con rhGH; sin embargo, en pacientes con osificaciones ectópicas el tratamiento con rhGH debe ser utilizado con precaución, dado que no hay datos accesibles acerca de su posible efecto en la evolución de las osificaciones |
| Alteraciones en la función gonadal |
|---|
| 3.18. La estadificación de maduración sexual de Tanner debe ser realizada en intervalos regulares para monitorizar la progresión de la pubertad en todos los pacientes con PHP y enfermedades relacionadas 3.19. Se deben evaluar el descenso y la localización testicular en varones con PHP o enfermedades relacionadas. La criptorquidia, cuando está presente, debe ser corregida y tratada de acuerdo con las recomendaciones estándar 3.20. No recomendamos el examen bioquímico rutinario del estado gonadal, a no ser que esté clínicamente indicado. El hipogonadismo, cuando está presente, debe ser tratado con hormonas sexuales siguiendo los mismos criterios estándar, dosis y seguimiento que cualquier otro tipo de hipogonadismo 3.21. Después de la pubertad, la historia menstrual debe ser recogida en cada visita de seguimiento, y en presencia de oligomenorrea o amenorrea en mujeres o síntomas de hipogonadismo en hombres se debe solicitar evaluación bioquímica 3.22. En caso de infertilidad, el tratamiento con reproducción asistida puede ser planteado de acuerdo con las guías nacionales 3.23. En pacientes con PHP y enfermedades relacionadas, las gestaciones naturales e inducidas deben ser vigiladas desde el punto de vista obstétrico de la misma forma que cualquier otra gestación. Sin embargo, la dosificación de las formas activas de vitaminaD y levotiroxina podría tener que ser ajustada. La posibilidad de parto por vía vaginal puede verse limitada como consecuencia de la reducción del tamaño de la pelvis y del margen de movimiento de las caderas debido a las osificaciones locales 3.24. El manejo de la hipocalcemia y el hipotiroidismo debe seguir las guías disponibles actuales de manejo de hipoparatiroidismo e hipotiroidismo durante el embarazo. En el recién nacido deberían evaluarse los niveles de TSH, calcio y fósforo. La lactancia no está contraindicada, pero se aconseja un estrecho seguimiento y monitorización clínica del niño (especialmente del peso) 3.25. Los pacientes con PHP y enfermedades relacionadas tienen diversos factores de riesgo potenciales para desarrollar osteoporosis (hipogonadismo, elevación crónica de la PTH y deficiencia de GH). Sin embargo, debido a la ausencia de evidencia de un aumento en el riesgo de fracturas, no existe indicación de realizar mediciones con DXA (dual-energy X-ray absorptiometry) de forma rutinaria en pacientes con PHP y enfermedades relacionadas. Si se diagnostica osteoporosis el manejo debe tener en cuenta, cuando sea posible, el tratamiento de la causa secundaria subyacente a la pérdida de hueso (hipogonadismo, estatus posmenopáusico o relacionada con la elevación mantenida de los niveles de PTH y deficiencia de GH) |
| Manejo de otras resistencias hormonales |
|---|
| 3.26. No recomendamos la medición rutinaria de calcitonina ni el cribado de resistencias hormonales adicionales en pacientes con PHP y enfermedades relacionadas |
| Obesidad y otros aspectos metabólicos |
|---|
| 3.27. Recomendamos la monitorización regular del IMC y del comportamiento alimentario. Los programas de educación, así como el apoyo psicológico, deben ser facilitados a los pacientes y familias cuando la obesidad y/o los trastornos alimentarios están presentes e incluso en presencia de un IMC normal, como estrategia preventiva, ya que estos pacientes tienen un alto riesgo. El consejo dietético debe tener en cuenta que estos pacientes tienen un menor gasto energético en reposo 3.28. Recomendamos que en todos los pacientes con PHP y enfermedades relacionadas sea evaluada la presencia de síntomas como trastornos del sueño, ronquido, déficit de atención y somnolencia diurna, que, en caso de presentarse, deben ser seguidos de la realización de polisomnografía 3.29. El metabolismo lipídico y de la glucosa debe ser monitorizado regularmente 3.30. La presión arterial debe ser monitorizada regularmente con un manguito de tamaño apropiado. Los pacientes con hipertensión deben ser tratados como esté indicado clínicamente |
| Manejo de las osificaciones ectópicas |
|---|
| 3.31. La presencia de placas cutáneas de hueso debe ser investigada mediante un examen cuidadoso en cada visita en todos los pacientes con mutaciones en GNAS, especialmente en aquellos con mutaciones en el alelo paterno (POH y PPHP). Los pacientes y sus familias deben ser instruidos sobre autoexploración. Recomendamos documentar lo siguiente en cada visita: localización y tamaño de las osificaciones; implicación de articulaciones y afectación del movimiento y crecimiento óseo; predilección de las lesiones por áreas expuestas a presión aumentada debido al peso soportado (pies y tobillos); valoración de los eventos desencadenantes (trauma, infección, inflamación y cirugía); asociación con el desarrollo puberal, y tratamiento con rhGH 3.32. No están indicadas las pruebas de imagen regulares para las osificaciones ectópicas 3.33. La evaluación por imagen de las osificaciones debe ser llevada a cabo utilizando TAC o RM, dependiendo de la localización, cuando las lesiones son dolorosas, sintomáticas, comprometen la función del órgano o se está considerando su resección quirúrgica 3.34. Cuando el diagnóstico de osificaciones ectópicas es dudoso y se ha excluido fibrodisplasia osificante progresiva, no hay contraindicación para realizar biopsia de la lesión, ya que ninguna evidencia sugiere que la inflamación o trauma lleve a una progresión de las osificaciones ectópicas en PHP y enfermedades relacionadas 3.35. La terapia física y el cuidado meticuloso de la piel son los enfoques más importantes para la prevención del desarrollo y/o progresión de las osificaciones ectópicas. La resección quirúrgica debe ser considerada en presencia de lesiones superficiales y delimitadas que asocien dolor y/o alteración funcional. El paciente debe ser referido a un cirujano con experiencia en el tratamiento de osificaciones ectópicas. En caso de osificaciones extensas alrededor de una articulación se debe evitar la inmovilización (mediante escayolas), ya que puede llevar a anquilosis de la articulación 3.36. No hay evidencia para recomendar el uso de fármacos antiinflamatorios no esteroideos, bifosfonatos o esteroides en el tratamiento primario o perioperatorio de las osificaciones ectópicas asintomáticas |
| Braquidactilia y aspectos ortopédicos |
|---|
| 3.37. Además de su uso para el diagnóstico y caracterización de la braquidactilia, la realización de pruebas radiológicas adicionales específicas se debe reservar para pacientes con sospecha clínica concreta de malformación ortopédica o alteración funcional, especialmente en presencia de signos o síntomas neurológicos. Los pacientes con braquidactilia severa deben ser evaluados formalmente en relación con sus capacidades motoras finas y ser provistos de aparatos ortopédicos adecuados cuando esté indicado (es decir, zapatos especiales y plantillas ortopédicas) 3.38. Se debe ofrecer evaluación y tratamiento específico multidisciplinar en caso de manifestaciones ortopédicas o neurológicas raras: estenosis espinal, deslizamiento bilateral de la epífisis de la cabeza femoral, anquilosis de la articulación temporomandibular, escoliosis precoz, malformación de Chiari tipo1 y sinostosis craneal |
| Manejo de la función cognitiva |
|---|
| 3.39. Considerar derivación a neuropsicología para evaluación neurocognitiva y/o conductual al diagnóstico o en edad preescolar, especialmente en pacientes con PHP1A y acrodisostosis producida por mutaciones en PDE4D, y también en el caso de que esté indicada (por ejemplo, si el paciente presenta síntomas de alteración cognitiva). La realización de pruebas adicionales y apoyo debe plantearse también cuando sea necesario |
| Riesgo de malignidad |
|---|
| 3.40. No hay datos para recomendar un cribado específico de cáncer en PHP y enfermedades relacionadas |
En muchas ocasiones la resistencia a la PTH se detecta al presentar el paciente signos causados por la presencia de hipocalcemia20. Esta hipocalcemia mantenida, asociada a la hiperfosfatemia, puede conllevar la aparición de calcificaciones intracraneales, así como depósitos de calcio/fósforo en los ojos, generando cataratas o degeneración macular37.
El objetivo del tratamiento frente a esta hipocalcemia debe ser algo más agresivo que el empleado en otros casos de hipoparatiroidismo, incluyendo el uso de metabolitos de la forma activa de la vitaminaD (calcitriol) o análogos (alfacalcidol), así como suplementos orales de calcio si fuese necesario, con el objetivo de normalizar los niveles de calcio. Es importante mantener los niveles séricos de PTH en la parte alta del rango de normalidad, evitando tanto la supresión de la PTH (lo que podría asociarse con hipercalciuria y desarrollo de nefrocalcinosis) como la presencia de hiperparatiroidismo (que asociaría efectos adversos en la mineralización esquelética o sobre la placa de crecimiento)2.
Crecimiento y deficiencia de hormona de crecimientoUn porcentaje importante de pacientes con PHP y enfermedades relacionadas presentan talla baja en su edad adulta. Así, en pacientes con PHP1A y PPHP la talla media final es de alrededor de 149cm en mujeres y de 155cm en hombres (−2,5SDS respecto a la población general)38. En pacientes con acrodisostosis esta desviación de la talla respecto a la población general es aún más marcada (−3,5SDS)26.
Esta afectación de la talla se va desarrollando con el tiempo. En estudios con cohortes pequeñas de pacientes con PHP1A se ha observado que la velocidad de crecimiento disminuye pocos años después del nacimiento, presentan un estirón puberal deficiente y un cese prematuro del crecimiento, junto con una aceleración de la maduración esquelética (más de +2SDS respecto a su edad cronológica), lo que, unido a disostosis (particularmente en el caso de la acrodisostosis), dificulta aún más la estimación del potencial patrón de crecimiento futuro39.
Los pacientes que concomitantemente con su PHP presentan deficiencia de hormona de crecimiento (GH) y/o pacientes nacidos con retraso del crecimiento intrauterino que no consiguen un crecimiento recuperador adecuado podrían beneficiarse del tratamiento con rhGH. Sin embargo, aún se necesitan más datos sobre la evolución del crecimiento en pacientes de estas características tratados con GH, así como respecto a la eventual utilidad del tratamiento en pacientes no deficientes de GH2.
Obesidad y comorbilidades metabólicasLa obesidad en pacientes con PHP1A parece ser la consecuencia de una combinación entre hiperfagia y gasto energético en reposo disminuido36. Conforme su edad progresa (infancia tardía, adolescencia y primera juventud), la hiperfagia suele disminuir y el gasto energético va ajustándose hacia la normalidad. Como consecuencia, la obesidad es menos marcada en la etapa adulta que en la infancia.
Esta obesidad temprana también es apreciable en pacientes con PHP1B. De hecho, estos pueden nacer ya con macrosomía o presentar una importante ganancia de peso en los primeros meses de vida.
Es, por tanto, imprescindible un estrecho control del peso (y patologías asociadas al exceso del mismo, como trastornos del sueño, alteraciones del metabolismo hidrocarbonado y lipídico, hipertensión) en todos los pacientes con alteraciones en el locus GNAS.
Desarrollo cognitivoLa adquisición de capacidades cognitivas puede estar alterada en pacientes con PHP1A6,23 y en pacientes con acrodisostosis causada por mutaciones en PDE4D18. En estos casos suele observarse un retraso en el desarrollo y problemas de aprendizaje, junto a un valor disminuido en la escala de inteligencia de Wechsler, y en el caso de los pacientes con PHP1A se ha descrito una incidencia aumentada de manifestaciones psiquiátricas que pudieran ser la consecuencia de una hipocalcemia mantenida durante largo tiempo.
Debería considerarse en ambos grupos de pacientes una valoración neuropsicológica al diagnóstico o en la edad preescolar para un estudio neurocognitivo y/o del comportamiento2.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
