





Since Albright and co-workers described pseudohypoparathyroidism in 1942 as the combined presence of hypocalcaemia and hyperphosphataemia associated with the existence of tissue resistance to parathyroid hormone (PTH) action upon normal renal function, great advances have been made in the clinical and genetic profile of patients affected by this condition. Furthermore, not only have genetic bases of pseudohypoparathyroidism been unravelled, but also variants in other genes involved in the PTH/PTHrP signalling pathway through Gsα, have been identified as the cause of diseases that share clinical features with pseudohypoparathyroidism.
In the paediatric setting, the first symptoms suggesting the impairment of this signalling pathway are the presence of subcutaneous ossifications, brachydactyly and/or early onset obesity, followed by the possible development of PTH resistance. This clinical suspicion should be confirmed by an accurate molecular diagnosis to allow for coordinated multidisciplinary clinical management. Among the features of this group of disorders, physicians should pay attention to evaluation of PTH and/or thyrotropin (TSH) resistance at diagnosis and throughout follow-up, as well as growth hormone deficiency, hypogonadism, skeletal deformities, dental impairment, obesity, insulin resistance, impaired glucose tolerance or type 2 diabetes mellitus and hypertension, as well as ectopic ossifications (either subcutaneous or affecting deeper tissues) and impairment of neurocognitive development.
Desde que en 1942 Albright y colaboradores describieran por primera vez el pseudohipoparatiroidismo como la existencia de hipocalcemia e hiperfosfatemia asociadas a resistencia tisular a la hormona paratiroidea (PTH) en presencia de una función renal normal, se han realizado grandes avances en la caracterización clínica y genética de los pacientes afectos de esta enfermedad. De hecho, no solo se han identificado las alteraciones moleculares responsables, sino que se ha podido establecer que variantes en otros genes de la misma vía de señalización, PTH/PTHrP a través de la proteína Gsα, son la causa de enfermedades que comparten determinadas manifestaciones clínicas con el pseudohipoparatiroidismo.
En el ámbito pediátrico, los primeros síntomas o signos que deben hacernos pensar en alteraciones en esta vía son la presencia de osificaciones subcutáneas, la braquidactilia y/o la obesidad de inicio precoz, seguidas en el tiempo por la posible aparición de resistencia a la PTH. Esta sospecha clínica deberá ser confirmada mediante un diagnóstico molecular que permita el correcto seguimiento clínico coordinado y multidisciplinar. Entre los aspectos a tener en cuenta en la atención de estos pacientes se incluye la evaluación al diagnóstico y seguimiento de la eventual presencia de resistencia a la PTH y a la hormona tirotropa (TSH), deficiencia de hormona de crecimiento (GH), hipogonadismo, alteraciones esqueléticas, alteraciones de la salud dental, obesidad, resistencia a la acción de la insulina, intolerancia a la glucosa o diabetes mellitus tipo 2 e hipertensión, así como osificaciones ectópicas (subcutáneas o con afectación de tejidos más profundos) y alteración del desarrollo neurocognitivo.
Pseudohypoparathyroidism (PHP) encompasses a group of diseases characterized by physical manifestations that usually include, with variable frequency, skeletal shortening, usually acromelic, short stature, early-onset obesity and ectopic ossifications in association with endocrine abnormalities that may include resistance parathyroid hormone (PTH) and thyroid stimulating hormone (TSH). These diseases are caused by genetic and/or epigenetic changes in the PTH/PTHrP signalling pathway involving the Gsα protein.1
This document is an abridged version of the first international consensus statement on the diagnosis and management of pseudohypoparathyroidism and related disease, recently published in the journal Nature Reviews Endocrinology,2 presenting the most relevant considerations and recommendations for the paediatric age group. We ought to highlight the participation of representatives of patient associations in the development of this consensus statement, including the one in Spain (asociacionpacientesphp@gmail.com).
Clinical diagnosisPseudohypoparathyroidism and related diseases comprehend a wide clinical spectrum. Some of its manifestations, such as ossifications, brachydactyly or hypocalcaemia, are frequently overlooked in very young children and eventually noticed as time goes by.
Case definitionsThe term “Albright's hereditary osteodystrophy” (AHO) is used to refer to a constellation of clinical manifestations first described by F. Albright that include a round face, a pyknic body type with short stature, brachydactyly (which may have late onset) and ectopic ossifications.3 Cognitive delay was subsequently added as another characteristic feature (although it is mainly found in patients with PHP1A); furthermore, obesity, especially with early onset, and macrocephaly relative to stature can also be part of the phenotype of AHO.4–6
Pseudohypoparathyroidism type Ia (PHP1A) was initially defined as the association of resistance to multiple hormones, including PTH and TSH, an AHO phenotype and reduced Gsα activity in in vitro assays.7 Resistance to TSH is usually detected early, in some cases during neonatal screening, while resistance to PTH tends to develop later on.8,9 Similarly, brachydactyly develops progressively, usually becoming evident before puberty.10 Other characteristics have since been described in patients with PHP1A (decreased neonatal length, early-onset obesity, cognitive disturbances). Thus, it is possible that patients with very early-onset obesity or with neonatal hypothyroidism actually have PHP1A but have not yet developed the classical clinical and chemical profile of the disease (resistance to PTH and/or brachydactyly), which may result in delayed diagnosis.2
Pseudohypoparathyroidism type Ic (PHP1C) has been defined as the association of all the features of PHP1A but with evidence of normal Gsα activity in in vitro complementation assays.11 Due to the overlap with PHP1A and the lack of assessment of Gsα activity in most published reports—and therefore the lack of differentiation between PHP1A and PHP1C—both the consensus statement and the summary we provide here do not specifically differentiate PHP1C, but instead treat it as a variant of PHP1A, both in the text and in the recommendations, unless otherwise specified.
Pseudohypoparathyroidism type Ib (PHP1B) was initially described as isolated resistance to PTH in the absence of the AHO phenotype and with normal levels of Gsα activity. Some patients with PHP1B have biochemical characteristics and some features of the AHO phenotype that overlap with PHP1A (which makes distinguishing these two diseases difficult).12,13
Pseudo-PHP (PPHP) is defined as presence of the AHO phenotype with reduced levels of Gsα activity in the absence of resistance to PTH.14 These patients often present with subcutaneous ossifications (osteoma cutis and bony plaques),15 and in some cases there is mild resistance to PTH and TSH.16
Progressive osseous heteroplasia (POH) consists in the presence of ectopic ossifications extending deeply into connective tissue. Patients with POH usually exhibit no other signs of AHO and normal sensitivity to PTH. The ossifications can cause severe ankylosis in the affected joints and local growth retardation.17
Acrodysostosis is defined as the association of severe brachydactyly, facial dysostosis and hypoplasia of the nasal root. The brachydactyly usually involves the phalanges, metacarpals and metatarsals excepting the thumbs and halluces. Radiographic examination reveals conical and accelerated bone maturation, and these changes may be present from birth or soon after.18
Pseudohypoparathyroidism type II is characterized by an increase in the levels of cAMP in response to the infusion of exogenous PTH, but with a deficient phosphaturic response.19 The molecular cause of this variant is still unknown.
Main clinical features of pseudohypoparathyroidism and related diseasesThe diagnosis of these diseases in the paediatric age group may be challenging, as many of their manifestations are nonspecific. The differential diagnosis of patients with obesity, early-onset hypothyroidism (possibly congenital), short stature and/or brachydactyly is very broad and includes multiple endocrine diseases and syndromes (Table 1).2
Differential diagnosis of pseudohypoparathyroidism (PHP) and related diseases based on the main presenting feature.
| Main feature | Differential diagnosis | Associated signs and comments |
|---|---|---|
| Hypocalcaemia with elevated PTH | Vitamin D deficiency or resistance | Improvement with vitamin D therapy. Rickets, alopecia. |
| Hypophosphatemic rickets | Enlargement of metaphyses. Genu valgum. Elevated alkaline phosphatase and hypophosphataemia. | |
| Hypoparathyroidism due to mutations in the PTH gene | Use different assays to confirm elevation of serum PTH levels. | |
| Brachydactyly | Trichorhinophalangeal syndrome due to mutations in the TRPS1 gene | Dysmorphism: slowly growing and sparse hair, laterally sparse eyebrows, bulbous nose, long and flat philtrum, thin upper lip and protruding ears. Hip dysplasia, small feet and shortening of first metatarsal, exostosis and ivory epiphyses. |
| Isolated brachydactyly type E associated with mutations in the HOXD13 gene | Syndactyly, lengthening of distal phalanges with shortening of the hallux. Hypoplasia/aplasia, lateral phalangeal duplication and/or clinodactyly. | |
| Brachydactyly mental retardation syndrome due to microdeletions in the 2q37 locus | Obesity, short stature. Brachydactyly. Cognitive impairment and abnormalities of psychomotor development. | |
| Turner syndrome (partial or complete absence of one of the X chromosomes) | Short stature, low birth weight, gonadal dysgenesis with ovarian failure, variable neurocognitive impairment. Brachydactyly and Madelung deformity. | |
| Brachydactyly type E with short stature due to mutations in the PTHLH gene | Short stature of variable severity, altered breast development. Oligodontia and delayed tooth eruption, malposition of teeth. Pseudoepiphysis. Brachydactyly | |
| Subcutaneous ossifications | Acne vulgaris | |
| Skin tumours such as pilomatrixomas, chondroid syringomas, basal cell carcinoma, pilar cysts and naevi Secondary or traumatic osteoma cutis and miliary osteoma | ||
| Inflammatory conditions, such as scars, chronic venous insufficiency, morphea, scleroderma, dermatomyositis or myositis ossificans progressiva | ||
| Progressive ossifications | Fibrodysplasia ossificans progressiva due to a recurrent activating missense mutation of ACVR1 | Progressive ossification of skeletal muscles, tendons, fascia and ligaments. Upper dorsal region and neck are regions affected first, trauma alters the natural course of disease. Congenital malformation of the great toes. |
| Tumoural calcinosis due to FGF23 or GALNT3 mutations | Deposition of calcium within the skin or muscles. Hyperphosphataemia. | |
| Early-onset obesity | Beckwith–Wiedemann syndrome | Hemihypertrophy. Macroglossia. |
| Genetic, cytogenetic or syndromic anomalies associated to early-onset obesity, such as Prader–Willi syndrome Monogenic causes of obesity (leptin-POMC signalling pathway, genes involved in pituitary axis development) | ||
| Early-onset hypothyroidism | Congenital hypothyroidism of any aetiology | Orthotopic, small-sized thyroid. Moderate TSH elevation. |
| Resistance to TSH due to TSH receptor mutations | ||
| Arterial hypertension | Autosomal dominant hypertension and brachydactyly type E syndrome | Short stature. |
PHP, pseudohypoparathyroidism; POMC, proopiomelanocortin; PTH, parathyroid hormone; TSH, thyroid-stimulating hormone.
This list of differential diagnoses is far from exhaustive, but considers the main diseases with presentations overlapping PHP and related diseases.
In most patients with PHP, the most prominent clinical manifestations are symptoms of hypocalcaemia secondary to resistance toPTH20 (usually with onset in late childhood).21 Periods of rapid growth (which entail an increase in calcium requirements) or nutritional calcium or vitamin D deficiency can trigger or exacerbate these symptoms.22
The first biochemical abnormalities observed in these patients are usually elevation of serum PTH and phosphate levels followed by hypocalcaemia (with low levels of calcium in urine and normal or low levels of calcitriol), with a gradual onset that lasts months or even years.23,24
Prolonged secondary hyperparathyroidism manifesting with chronic hypocalcaemia and 1,25-dihydroxyvitamin D deficiency has been associated with the development of tertiary hyperparathyroidism in adult patients with PHP1B25 (although it may also develop in adolescents) and can cause bone resorption and demineralization similar to those observed in rickets.
Resistance to PTH is also found in patients with acrodysostosis secondary to mutations in the PRKAR1A gene, although the presence of hypocalcaemia has not been documented in this group. Patients with mutations in the PDE4D gene usually have normal levels of PTH.26
Ectopic ossification may occur in diseases caused by changes in the GNAS gene, such as PHP1A/1C, PPHP and POH. Ectopic ossification is not the same as calcification and is not related to serum levels of calcium or phosphorus. It may manifest in the form of plaques (osteoma cutis) at birth or in the early years of life, or as isolated nodules in older children. There may be multiple nodules, and the nodules are hard on palpation and of variable size.27,28 There is no evidence that ossifications in these patients are the result of local traumatic injury. Ectopic ossifications are found in 100% of patients with POH, 80–100% of patients with PPHP/AHO, 30–60% of patients with PHP1A and in exceptional cases of PHP1B.23 To date, there are no reports of ossifications in patients with acrodysostosis. In patients with POH, ossifications develop gradually and extend into the muscles, tendons and ligaments. They may be limited or be more prominent in one side of the body.17
Brachydactyly in PHP and related diseases can be classified as type E, which is defined as a variable shortening of the metacarpals (especially the 5th, 4th and 3rd metacarpals), usually combined with normal length of phalanges and occasionally by relative shortening of the metatarsals. Brachydactyly develops over time and may not be present in the early stages of life,10,29 except in patients with acrodysostosis,18 and its frequency and severity vary between the different diseases in this group (with the highest prevalence being 100% in patients with acrodysostosis). In any case, brachydactyly is not specific to PHP and related disorders and can be found in patients with many other disorders30 (Table 1).
Patients with PHP1A frequently (if not always) exhibit elevated serum levels of TSH and normal or mildly decreased levels of thyroid hormones. Some patients present with clinically significant hypothyroidism. The elevation of TSH due to resistance to this hormone may be present since birth and detected during neonatal screens. In PHP1B, TSH levels are in the upper limit of normal or mildly elevated in 30% to 100% of patients.2,23,31 Resistance to TSH may be found in patients with acrodysostosis due to PRKAR1A mutations, but not in patients with PDE4D mutations.26,32 A large subset of patients with PHP1A and PHP1B have elevated levels of calcitonin, while resistance to gonadotropins seems to be considerably milder compared to resistance to other hormones (such as TSH or PTH), which has led to the hypothesis that patients with PHP1A have only partial resistance to gonadotropins.2
There are other signs that, on account of not causing measurable hormone changes, may not be recognized as manifestations of the spectrum of PHP and related diseases. In fact, while these early symptoms may already be apparent in early childhood, the diagnosis is often delayed until puberty in patients with PHP1A or acrodysostosis, and even to adulthood in patients with PHP1B, unless the patient has a positive family history. The presence of ossifications may lead to earlier diagnosis in cases of POH or osteoma cutis. Among these changes, we ought to highlight anthropometric abnormalities in newborns and obesity. The presence of small anthropometric values for gestational age has been described in patients with mutations in the paternal GNAS allele33 and in most patients with acrodysostosis and mutations in the PRKAR1A or PDE4D genes.26,32 Most of these patients have short stature as adults (the mean final height z-score in patients with PPHP or AHO is of less than −2.0), while patients with methylation changes in the GNAS locus exhibit moderately increased birth weights and lengths, and, while the data on their adult height is scarce, they suggest that the final height is within the normal range.34
Although it is also a nonspecific feature, the development of obesity is associated with PHP-related diseases, such as PHP1A/1C or acrodysostosis. We ought to note that obesity in these patients is more pronounced in childhood compared to adulthood. Patients with PHP1A may also exhibit a progressive reduction in insulin sensitivity.5,35,36
We recommend that clinicians be particularly mindful of these manifestations, since accurate diagnosis can have extremely important implications in management, as it allows: (a) early screening and treatment of associated endocrine complications, such as TSH or PTH resistance; (b) prevention and treatment of obesity or short stature; (c) adequate treatment of ectopic ossifications and (d) adequate genetic and prenatal counselling.
Evolution of the classification of pseudohypoparathyroidism and related diseasesThe current classification distinguishes variants of PHP such as PHP1A, PHP1B, PHP1C and PPHP. As we mentioned above, classical subtype assignment is based on the presence of the AHO phenotype and the characterization of hormone resistance patterns and Gsα protein activity through in vitro assays. However, this scheme excludes diseases like acrodysostosis or POH, whose clinical phenotypes may overlap. This overlap poses a challenge to diagnosis and renders the current classification scheme obsolete. Furthermore, the tools available for assessment have evolved, the technique to measure Gsα activity in cell membranes is not widely available, and molecular diagnosis has become the gold standard for differentiation of PHP variants. While a new classification scheme has been proposed for this set of diseases,1 it remains to be validated.
Molecular diagnosisFig. 1 presents a sequential algorithm with the recommendations for the molecular assessment and diagnosis of PHP and related diseases.
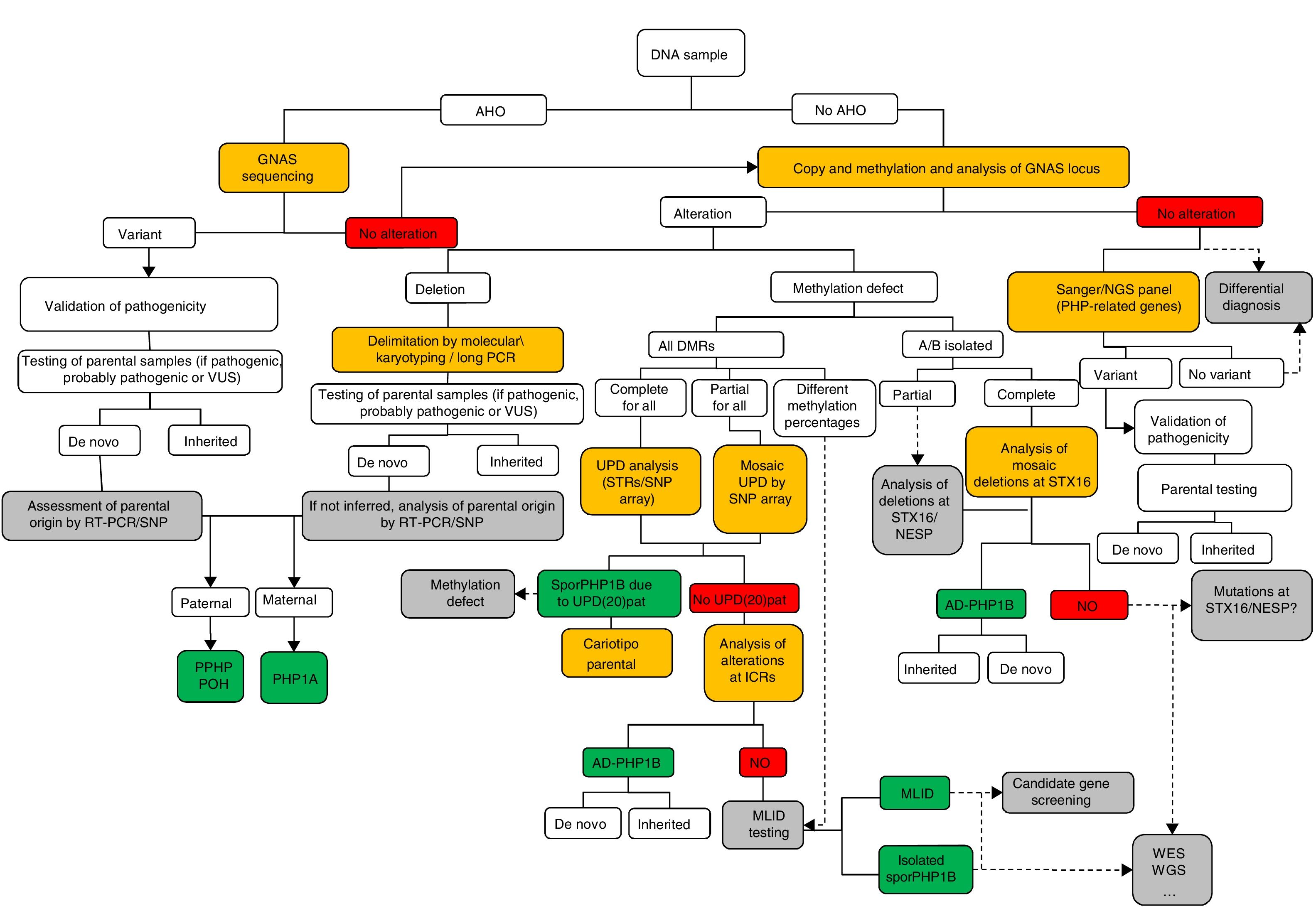
Molecular algorithm for the confirmation of diagnosis of PHP and related disorders.
If the patient presents with Albright hereditary osteodystrophy (AHO), genetic alterations at GNAS should be assessed, including point mutations (sequencing) and genomic rearrangements. Once the variant is found, its pathogenicity should be confirmed according to guidelines40, and, when possible, the parental origin should be determined.
In the absence of AHO, the investigation should start with the assessment of epigenetic alterations. Based on the results of the investigation of the methylation status, further tests will be performed to reach the final diagnosis: (a) if the methylation defect is restricted to GNAS A/B:TSS-DMR, STX16 deletions should be screened for, and, if present, the diagnosis of autosomal dominant-pseudohypoparathyroidism type 1B (AD-PHP1B) is confirmed; (b) if methylation is abnormal at the four DMRs, paternal uniparental disomy of chromosome 20 (UPD(20q)pat) should be screened for; (c) in the absence of UPD(20q)pat, deletions at NESP/AS should be screened for; (d) if no genetic cause is identified as the cause of the methylation defect, the sporadic form of the disease (sporPHP1B) is suspected.
After exclusion of the GNAS locus as the cause of the phenotype, and in patients with AHO, pseudohypoparathyroidism (PHP)-related genes should be sequenced (that is, at least PDE4D and PRKAR1A).
Squares in yellow indicate the technology; in green, the final molecular confirmation; and in grey, suggested future steps or research.
ICRs, imprinting control regions; MLID, multi-locus imprinting disturbance; NGS, next-generation sequencing; POH, progressive osseous heteroplasia; RT-PCR, reverse-transcription PCR; SNP, single-nucleotide polymorphism; STRs, short tandem repeats (microsatellites); UPD, uniparental disomy; VUS, variant of uncertain significance; WES, whole exome sequencing; WGS, whole genome sequencing.
In case of clinical suspicion of PHP, it is recommended that the molecular diagnosis include DNA sequence, methylation and copy number variant analyses at the GNAS locus.2
In the paediatric age group, patients are most frequently diagnosed after displaying some of the characteristics of the AHO phenotype (mainly short stature and/or early-onset obesity), so the molecular evaluation should start with the sequencing of the GNAS gene.21 A similar approach should be applied to the investigation of patients with ectopic ossifications.2 If the causative variant has been identified, it is important to determine whether it is located in the paternal or the maternal allele in order to plan the appropriate followup. This can be done by genetic testing of the parents or, in case of de novo mutations, by RT-PCR or SNP analysis.
In the absence of manifestations of AHO, patients are usually evaluated to assess the symptoms of hypocalcaemia,21 and the molecular evaluation should start with the determination of the level of methylation at the GNAS locus. When changes in methylation are detected, the underlying genetic causes should be investigated (deletions in STX16, NESP/AS or uniparental disomy) for the purpose of providing adequate genetic counselling.
In cases of severe brachydactyly involving every bone in the hand and with onset in the early years of life, it is important to rule out acrodysostosis by sequencing the PRKAR1A and PDE4D genes.26
Treatment and followupAt presence, there is no evidence from prospective clinical trials focused on the management and followup of patients with PHP and related diseases. The recommendations given in Table 2 are the result of a review of the literature published until the consensus process and the expert opinion of its participants. Although a comprehensive review of the recommendations for treatment and followup can be found in Table 3, we will devote a few paragraphs to summarize the most important aspects of the management of paediatric patients.
Summary of the main interventions to perform in paediatrics practice for the followup of patients with pseudohypoparathyroidism and related diseases.
| Birth to age 2 years | Early childhood, 2–6 years | Late childhood-adolescence | |
|---|---|---|---|
| Anticipatory guidance | |||
| Family counselling | • | • | • |
| Genetic counselling | At diagnosis | At diagnosis | At diagnosis |
| Medical evaluation | |||
| Linear growth | • | • | • |
| Weight gain/BMI | • | • | • |
| Descent of testes | • | • | If not assessed before |
| Blood pressure | N/A | •* | • |
| Development/cognition | • | • | S |
| Psychosocial evaluation | N/A | • | S |
| Ectopic ossifications | • | • | |
| Orthodontic/dental | N/A | • | • |
| Bone age study | N/A | • (in case of growth deceleration) | • (in case of growth deceleration) |
| Calcium and phosphate homeostasis | • | • | • |
| Thyroid | • | • | • |
| Puberty | N/A | N/A | (biochemistry tests if delayed puberty) |
| GH/IGF axis | N/A | • | • |
| Carbohydrate and lipid metabolism | N/A | • | • |
| Fertility | N/A | N/A | S |
• At diagnosis and subsequently once a year; N/A, not applicable; S, subjective (based on medical history and physical examination).
Recommendations for the management and followup of patients with pseudohypoparathyroidism and related diseases.
| Treatment and care |
|---|
| 3.1. The diagnosis and management of patients with PHP and related diseases should be performed by a multidisciplinary team of specialists. |
| Management of resistance to PTH |
|---|
| 3.2. At diagnosis or before starting treatment, we recommend monitoring of serum levels of PTH, calcium, phosphorus and 25-hydroxyvitamin D. Measurement of PTH, calcium and phosphorus should be performed regularly (every 6 months in children and at least yearly in adults) with the exception of patients carrying either a GNAS mutation on the paternal allele or a PDE4D mutation in whom (apart from at diagnosis) routine assessment is not necessary. Monitoring of serum levels of calcium should be more frequent in symptomatic individuals, during acute phases of growth, during acute illness and during pregnancy and breastfeeding, when dose requirements for active vitamin D metabolites or analogues might change. Levels of 25-hydroxyvitamin D should be normalized and maintained in the normal range in all patients. 3.3. Severe symptomatic hypocalcaemia requires immediate correction, which may include intravenous administration of calcium salts according to general guidelines for the management of acute hypocalcaemia in hypoparathyroidism. These patients should also receive concomitant treatment with active vitamin D metabolites (calcitriol) or analogues (alfacalcidol). 3.4. In patients with substantial and progressive increases in levels of PTH and hyperphosphataemia, treatment with active vitamin D metabolites or analogues could be considered, independently of the presence of hypocalcaemia. Calcium supplements should be considered depending on the dietary calcium intake. Serum levels of phosphorus should be monitored during treatment with vitamin D metabolites or analogues and calcium supplements. 3.4.b. Treatment with active vitamin D metabolites or analogues can also be considered when levels of PTH are more than double the upper level of normal, independently of the presence of hypocalcaemia. 3.5. The objectives of conventional management of PTH resistance include maintenance of serum calcium and phosphorus levels within the normal range while avoiding hypercalciuria (corrected for age and size) and lowering PTH levels as permitted by serum and urinary levels of calcium. We recommend the use of the active vitamin D metabolite calcitriol or the active vitamin D analogue (alfacalcidol) with or without calcium supplementation as the mainstay of treatment of chronic hypocalcaemia. Patients should not be treated with PTH or PTH analogues. During treatment, levels of PTH, calcium and phosphorus should be monitored every 6 months in asymptomatic patients and more frequently when clinically indicated. Patients and/or their families should be educated about the signs and symptoms of hypocalcaemia and hypercalcaemia. 3.6. We recommend the use of appropriate imaging tests to assess for the presence of nephrocalcinosis during the transition to adulthood. 3.7. We recommend age-appropriate imaging tests to monitor the development or worsening of nephrocalcinosis in patients with persistent hypercalciuria on repeated measurements and as clinically indicated. 3.8. A brain CT scan is indicated for the evaluation of long-term consequences of hypocalcaemia and hyperphosphataemia only when neurological manifestations are present. An ophthalmological examination is recommended to diagnose or exclude cataracts. 3.9. Phosphate binders (other than calcium) are rarely, if ever, indicated in the management of severe and long-term persistent hyperphosphataemia. 3.10. We recommend regular dental checkups every 6–12 months during childhood and as clinically indicated in adults. |
| Management of resistance to TSH |
|---|
| 3.11. Evaluation of thyroid function (including autoantibodies in adolescents and adults) for early detection of TSH resistance and early intervention is recommended in all patients with PHP and related disorders at diagnosis. Thereafter, TSH monitoring is recommended every 6 months in patients aged <5 years and yearly in older children and adults. 3.12. The indication or indications to treat hypothyroidism, the dosage of levothyroxine and the therapeutic goals should be the same as for any patient with hypothyroidism or subclinical hypothyroidism. |
| Management of growth disturbances and GH deficiency |
|---|
| 3.13. We recommend careful monitoring of height in children at every checkup (at least every 6 months) until final height is reached. 3.14. We recommend monitoring of skeletal maturation using plain radiography in all children (with the exception of those with PHP1B) and evaluation for GH deficiency in all children exhibiting linear growth deceleration. As most patients develop GH deficiency due to GHRH resistance, clinical and/or biochemical evaluation of the GH–insulin-like growth factor 1 (IGF1) axis should be performed in all patients, typically around the age of 3–6 years and subsequently repeated as needed. 3.15. Patients with GH deficiency should be treated with rhGH. Data are needed on the outcomes of GH treatment in children who are not GH-deficient before recommending this treatment in these patients as well. 3.16. Treatment with rhGH may be considered in adults with GH deficiency; however, specific evidence of the benefits of rhGH in this population is lacking, and treatment should be given according to country-specific regulations. 3.17. Patients with PHP who were born SGA and do not exhibit appropriate catch-up growth might be eligible for treatment with rhGH; however, caution should be exercised in patients with ectopic ossifications, as no data are available on the possible effect of rhGH treatment on the outcome of ossifications. |
| Alterations of gonadal function |
|---|
| 3.18. Tanner staging of sexual maturation should be performed at regular intervals to monitor pubertal progression in all patients with PHP or related disorders. 3.19. Testicular descent and location should be assessed in males with PHP or related disorders. Cryptorchidism, when present, should be corrected and managed according to the standard recommendations. 3.20. We do not recommend routine biochemical assessment of the gonadal status unless clinically indicated. Hypogonadism, when present, should be treated with sex hormones following the same standard criteria, dosage and followup as in any other form of hypogonadism. 3.21. After puberty, menstrual history should be collected at each follow-up visit, and a biochemical evaluation should be requested in the presence of oligomenorrhoea or amenorrhoea in women and of hypogonadal symptoms in men. 3.22. In case of infertility, assisted reproductive treatment can be considered in adherence to national guidelines. 3.23. In patients with PHP and related disorders, natural and induced pregnancies should be monitored from an obstetrical point of view in the same way as any other pregnancy. However, dosages of active forms of vitamin D and levothyroxine might have to be adjusted. The possibility of vaginal delivery might be limited as a result of reduced pelvic size and decreased range of motion of the hips due to local ossifications. 3.24. Management of hypocalcaemia and hypothyroidism should follow the current guidelines for the management of hypoparathyroidism and hypothyroidism during pregnancy. The levels of TSH, calcium and phosphorus should be measured in the newborn. Breastfeeding is not contraindicated, but close followup and clinical monitoring of the infant (particularly of weight) is recommended. 3.25. Patients with PHP and related disorders have several potential risk factors for osteoporosis (hypogonadism, chronic elevation of PTH and GH deficiency). However, owing to the lack of evidence of increased fracture risk, routine performance of dual-energy X-ray absorptiometry (DXA) measurements is not indicated in patients with PHP and related disorders. If osteoporosis is diagnosed, management should take into account, whenever possible, treatment of the underlying secondary cause of bone loss (hypogonadism, postmenopausal status or cause related to sustained elevation of PTH levels and GH deficiency). |
| Management of other hormone resistances |
|---|
| 3.26. We do not recommend routine calcitonin measurement or screening for additional hormone resistances in patients with PHP and related disorders. |
| Obesity and other metabolic issues |
|---|
| 3.27. We recommend regular monitoring of BMI and eating behaviour. Educational programmes, as well as psychological support, should be provided to patients and families when obesity and/or eating disorders are present and even in the presence of a normal BMI as a preventive strategy, as these patients are at high risk. Nutritional counselling should take into account that these patients have a decreased resting energy expenditure. 3.28. We recommend that all patients with PHP and related disorders be evaluated for symptoms such as restless sleep, snoring, inattentiveness and daytime somnolence, with performance of polysomnography if symptoms are present. 3.29. Lipid and glucose metabolism should be monitored on a regular basis. 3.30. Blood pressure should be monitored regularly with an appropriately sized cuff. Patients with hypertension should be treated as clinically indicated. |
| Management of ectopic ossifications |
|---|
| 3.31. The presence of cutaneous bony plaques should be investigated by careful examination at each visit in all patients with GNAS mutations, especially those with mutations on the paternal allele (POH and PPHP). Patients and families should be instructed about self-examination. We recommend documenting the following at each visit: location and size of ossifications; involvement of joints and impairment of movement and bone growth; predominance of lesions in areas exposed to increased pressure due to weight bearing (feet and ankles); assessment of triggering events (trauma, infection, inflammation and surgery); association with pubertal development; and treatment with rhGH. 3.32. Regular imaging of ectopic ossifications is not indicated. 3.33. Imaging of ossifications should be performed using CT or MRI, depending on localization, when the lesions are painful, symptomatic, jeopardize joint or organ function or are being considered for surgical excision. 3.34. When the diagnosis of ectopic ossifications is doubtful and when fibrodysplasia ossificans progressiva has been excluded, there is no contraindication to perform a biopsy of the lesion, as no evidence suggests that inflammation or trauma leads to progression of ectopic ossifications in PHP and related disorders. 3.35. Physical therapy and meticulous skin care are the most important strategies for the prevention of development and/or progression of ectopic ossifications. Surgical excision should be considered in the presence of delimited, superficial lesions associated with pain and/or functional impairment. The patient should be referred to a surgeon with experience in the management of ectopic ossifications. In extensive ossifications around a joint, avoid immobilization (through casts), as this might lead to ankylosis of the joint. 3.36. There is no evidence for recommending the use of nonsteroidal anti-inflammatory drugs, bisphosphonates or steroids in the primary or peri-surgical treatment of asymptomatic ectopic ossifications. |
| Brachydactyly and orthopaedic issues |
|---|
| 3.37. In addition to their use for diagnosis and characterization of brachydactyly, further specific radiological investigations should be reserved for patients with specific clinical suspicion of orthopaedic malformation or functional impairment, particularly in the presence of neurological signs or symptoms. Patients with severe brachydactyly should undergo a formal evaluation of their fine motor skills and be supplied with appropriate orthopaedic devices when indicated (that is, special shoes and orthopaedic insoles). 3.38. Specific multidisciplinary evaluation and therapy should be offered for rare orthopaedic or neurological manifestations: spinal stenosis, bilateral slipped capital femoral epiphyses, temporomandibular joint ankyloses, precocious scoliosis, Chiari malformation type 1 and cranial synostosis. |
| Management of cognitive function |
|---|
| 3.39. Consider referral to a neuropsychologist for neurocognitive and/or behavioural assessment at diagnosis or in preschool age, particularly in patients with PHP1A and acrodysostosis due to PDE4D mutations, and in any other case where it is appropriate (for instance, if the patient exhibits symptoms of cognitive impairment). Consider additional testing and support as needed. |
| Risk of malignancy |
|---|
| 3.40. There are no data to recommend a specific screening for malignancies in PHP and related disorders. |
In many cases, resistance to PTH is identified when the patient develops manifestations of hypocalcaemia.20 This chronic hypocalcaemia, in association with hypophosphataemia, can result in the intracranial deposition of calcium and the deposition of calcium and phosphorus in the eyes, which may lead to the development of cataracts or macular degeneration.37
The approach to the treatment in patients with chronic hypocalcaemia must be more aggressive compared to other types of hypoparathyroidism, including administration of active vitamin D (calcitriol) or analogues (alfacalcidol), as well as oral calcium supplements as needed to normalize serum calcium levels. It is important to maintain serum levels of PTH in the upper range of normal, avoiding both suppression of PTH (which would result in hypercalciuria and the development of nephrocalcinosis) and the development of hyperparathyroidism (which would have an adverse effect on bone mineralization or on growth plates).2
Management of growth and growth hormone deficiencyA considerable percentage of patients with PHP and associated diseases have short stature in adulthood. Thus, the mean final height of patients with PHP1A and PPHP is about 149cm in women and 155cm in men (2.5 SDs below the general population).38 In patients with acrodysostosis the deviation from the general population is even more pronounced (z, −3.5).26
Short stature develops over time. Small cohort studies in patients with PHP1A have found that growth velocity slows down a few years after birth, with a decreased pubertal growth spurt, early growth cessation and accelerated bone maturation (bone age 2 SDs greater than chronological age), which, combined with dysostosis (especially in patients with acrodysostosis) further complicates the prediction of future growth.39
Patients in whom PHP is associated with growth hormone (GH) deficiency and/or patients with a history of intrauterine growth restriction that do not exhibit adequate catch-up growth could benefit from treatment with recombinant human GH (rhGH). However, we need further data on outcomes of treatment with rhGH in patients with these characteristics and also on the potential usefulness of this treatment in patients without GH deficiency.2
Obesity and metabolic comorbiditiesObesity in patients with PHP1A seems to result from a combination of hyperphagia and a decreased resting energy expenditure.36 As age increases (late childhood, adolescence and early youth), hyperphagia usually decreases while the energy expenditure gradually normalizes. Consequently, obesity is less pronounced in adulthood compared to childhood.
This early obesity is also observed in patients with PHP1B. In fact, they may already be born with macrosomia or exhibit significant weight gains in the first months of life.
Consequently, it is essential to closely monitor weight (and diseases associated with excess weight, such as sleep disorders, disorders of carbohydrate or lipid metabolism, hypertension) in all patients with changes in the GNAS locus.
Cognitive developmentCognitive development may be altered in patients with PHP1A6,23 and patients with acrodysostosis secondary to mutations in PDE4D.18 These patients usually exhibit developmental delays and learning problems associated with low scores in the Wechsler intelligence scale, and there is evidence that patients with PHP1A develop psychiatric manifestations more frequently, possibly as a result of long-term hypocalcaemia.
In both groups of patients, performance of a neuropsychiatric evaluation should be considered at the time of diagnosis or in early childhood to assess neurocognitive development and/or behaviour.2
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Martos-Moreno GÁ, Lecumberri B, Pérez de Nanclares G. Implicaciones en pediatría del primer consenso internacional para el diagnóstico y asistencia a pacientes con pseudohipoparatiroidismo y enfermedades relacionadas. An Pediatr (Barc). 2019;90:125.e1–125.e12.
