






La hipoplasia pulmonar es la anomalía congénita más frecuentemente asociada a mortalidad perinatal.
Material y métodosSe ha realizado un estudio descriptivo retrospectivo de los casos de hipoplasia pulmonar diagnosticados entre 1995 y 2014 en un hospital universitario de tercer nivel, analizando estudios prenatales, manifestaciones clínicas, pruebas diagnósticas, tratamiento, datos de seguimiento a largo plazo y supervivencia.
ResultadosSe identificaron 60 casos, todos con estudio prenatal. Dieciséis recibieron intervención quirúrgica intraútero. La hernia diafragmática congénita fue la entidad más frecuentemente asociada. La manifestación clínica más habitual fue distrés respiratorio neonatal, hipoxemia grave y necesidad de soporte con ventilación mecánica. Se halló un 47% de mortalidad antes de los 60 días de vida y del 75% en las primeras 24 h de vida. Durante el seguimiento de los supervivientes se detectaron episodios de neumonías y bronquitis recidivantes, función pulmonar con patrón obstructivo y aceptable calidad de vida y tolerancia al ejercicio.
ConclusionesLa elevada mortalidad neonatal y la importante morbilidad a largo plazo de la hipoplasia pulmonar requieren de un diagnóstico temprano y la intervención de un equipo multidisciplinar especializado.
Pulmonary hypoplasia is the most frequent congenital anomaly associated with perinatal mortality.
Material and methodsA retrospective and descriptive review was conducted on cases of patients diagnosed with pulmonary hypoplasia between 1995 and 2014 in a tertiary university hospital. An analysis was made of the prenatal imaging, clinical manifestations, post-natal diagnostic tests, treatment and management, long-term follow up, and survival data.
ResultsA total of 60 cases were identified, all of them with prenatal imaging. Sixteen patients required foetal surgery. Congenital diaphragmatic hernia was the most frequent diagnosis. Main clinical presentation was respiratory distress with severe hypoxemia and high requirements of mechanical ventilation. Mortality rate was 47% within first 60 days of life, and 75% for the first day of life. Pneumonia and recurrent bronchitis episodes were observed during follow-up. They had a lung function obstructive pattern, and their quality of life and exercise tolerance was good.
ConclusionsHigh neonatal mortality and significant long-term morbidity associated with pulmonary hypoplasia requires an early diagnosis and a specialised multidisciplinary team management.
La hipoplasia pulmonar (HP) es una alteración congénita caracterizada por el escaso crecimiento y desarrollo del parénquima pulmonar, la vía aérea y los vasos pulmonares1-5. Su incidencia en la población general es de 9 a 11 casos por cada 10.000 nacimientos vivos2,3, aunque está subestimada debido a casos no diagnosticados con grados menores de HP que sobreviven al periodo neonatal2. En series de autopsias perinatales la prevalencia varía entre 7,8 y 26%1,3, siendo la anomalía más frecuentemente asociada a mortalidad perinatal1-6.
La causa más frecuente de HP es la secundaria al desequilibrio entre la cavidad torácica, los movimientos respiratorios fetales, la presión positiva del líquido pulmonar fetal y el volumen normal del líquido amniótico1-7. En la tabla 1 se han recogido y se enumeran las entidades más conocidas que se asocian a HP4,8,9. Las manifestaciones clínicas dependen del grado de afectación pulmonar: desde formas graves bilaterales a formas más leves unilaterales o lobares1,10. En el periodo neonatal puede presentarse con insuficiencia respiratoria, hipertensión pulmonar o hemorragia pulmonar. Es característica la insuficiencia respiratoria de inicio súbito, requiriendo ventilación mecánica con presiones elevadas en ausencia de atelectasias y obstrucciones2,11.
Clasificación de posibles mecanismos que originan hipoplasia pulmonar secundaria
| Alteraciones de la cavidad torácica | Alteración de la circulación del líquido pulmonar fetal | Alteración de los movimientos respiratorios fetales |
|---|---|---|
| Lesiones ocupantes de espacio Hernia congénita Diafragmática Malformación de la vía aérea (p. ej., malformación adenomatoidea quística) Derrames pleurales Onfaloceles Gastrosquisis | Hipoplasia de cavidades derechas o izquierdas cardiacas Estenosis pulmonar Oligohidramnios Agenesia renal Displasia quística Obstrucción del flujo urinario Rotura prolongada de membranas Hidropesía fetal | Desórdenes neuromusculares Problemas neurológicos Síndrome de Pena-Shokeir |
| Malformaciones de la pared torácica | ||
| Cifoescoliosis | ||
| Displasias esqueléticas |
En la etapa prenatal el diagnóstico de sospecha se basa en la ecografía tridimensional y la resonancia magnética (RM), que permiten determinar el volumen pulmonar12,13. En la etapa posnatal se usa la tomografía computarizada (TC) torácica y la gammagrafía de perfusión pulmonar, y la confirmación se hace mediante el estudio anatomopatológico1-4.
Existen distintos tipos de intervención quirúrgica prenatal para paliar el grado de HP en el feto, como el uso de drenajes en casos de hidrotórax o la oclusión traqueal fetoscópica con globo endotraqueal (del inglés fetoscopic tracheal occlusion [FETO]) en casos graves de hernia diafragmática congénita14,15. En la etapa posnatal, el tratamiento consiste en garantizar el soporte que permita una adecuada oxigenación mientras se logra el crecimiento y desarrollo pulmonar.
En el caso de la hernia congénita diafragmática se ha establecido una estrategia de tratamiento que consiste en la reparación quirúrgica posnatal y el uso de técnicas de soporte de ventilación invasiva que permitan cierta hipercapnia, además de un tratamiento intensivo contra la hipertensión pulmonar10,15-18.
La optimización del diagnóstico, los tratamientos y los cuidados posnatales de los pacientes afectados de HP es motivo actualmente de distintos estudios19. El objetivo de nuestro trabajo es describir las causas, los métodos diagnósticos y la evolución clínica de los pacientes afectados de HP diagnosticados en un hospital universitario de tercer nivel.
Material y métodosSe ha realizado un estudio descriptivo retrospectivo de los casos de HP diagnosticados entre 1995-2014 en el Hospital Universitario Vall d’Hebron de Barcelona, que fueron atendidos por los servicios de obstetricia, neonatología, cirugía pediátrica y neumología pediátrica.
Las variables analizadas fueron: edad gestacional del feto al diagnóstico, anomalías en ecografía prenatal, intervención quirúrgica prenatal, edad gestacional al nacimiento, edad al comienzo de la clínica respiratoria, sintomatología presentada, diagnóstico etiológico, pruebas diagnósticas utilizadas, resultados anatomo-patológicos, lateralidad de la afección pulmonar, tiempo requerido de ventilación mecánica y de oxigenoterapia, evolución clínica respiratoria según presencia de infecciones y sibilancias recurrentes, evolución de la función pulmonar, calidad de vida y supervivencia.
El tipo de intervenciones quirúrgicas prenatales estudiadas fueron: colocación de drenajes toraco-amnióticos en casos de hidrotórax y técnica FETO en casos de hernia diafragmática congénita que cumplían los criterios de edad gestacional menor o igual de 26 semanas y marcadores de alta mortalidad, como un índice pulmonar cefálico (del inglés lung head ratio [LHR]) inferior a 1 o herniación hepática a la cavidad torácica.
El diagnóstico de HP se basó en la gammagrafía pulmonar de perfusión, TC torácica y en criterios clínicos. El diagnóstico mediante gammagrafía se hizo detectando disminución de la perfusión pulmonar y mediante TC detectando disminución del volumen pulmonar y de los vasos pulmonares del lado afectado respecto a los contralaterales. El diagnóstico clínico se basó en la presencia de distrés respiratorio neonatal, requerimiento de ventilación mecánica con presiones elevadas y radiografía simple de tórax con escasa aireación y campos pulmonares pequeños.
Se realizó exploración funcional respiratoria en niños mayores de 5 años colaboradores, espirometría forzada, pletismografía y medición de la difusión (single breath [DLCO]), según estándares de la European Respiratory Society (ERS). Para ello se utilizó un espirómetro Jaeger® y un pletismógrafo Jaeger Masterscreen™ (CareFusion, San Diego, EE. UU.). Los datos se expresaron como Z-scores ajustados para el sexo, la edad y la talla según ecuaciones de la Global Lung Initiative (GLI) 2012 y Rosenthal, respectivamente.
Para describir la calidad de vida se revisaron los datos de historia clínica de visitas anuales como: escolaridad, tipo de actividad física realizada y limitación de esta comparada con niños de su misma edad normales. Se ha analizado la supervivencia mediante el método Kaplan-Meier en el programa SPSS versión 21.
ResultadosSe hallaron 60 casos, 33 varones (55%). Treinta y cinco casos presentaron afectación unilateral (21 izquierda-15 derecha) y 24 bilateral. Todos los pacientes fueron estudiados en el periodo prenatal mediante ecografía de 3 dimensiones; 46 pacientes mostraron alteraciones en ecografías prenatales: 19 oligoamnios, 21 hernia diafragmática, 6 hidrotórax, 2 displasia torácica, uno eventración diafragmática y uno polihidramnios. La media de edad gestacional del primer hallazgo ecográfico fue de 27 semanas (rango 15-39 semanas de gestación). En 7 pacientes se practicó una RM intraútero: en 6 casos se confirmaron la presencia de hernia congénita diafragmática y la disminución del tamaño del pulmón ipsolateral, y en un caso se detectó artrogriposis y disminución del tamaño pulmonar bilateral.
Cuatro pacientes fueron catalogados de HP primaria y 56 pacientes de HP secundaria, siendo la hernia diafragmática congénita (41%) la causa más frecuente de HP. La etiología hallada se describe en la tabla 2. Los 19 casos que presentaron oligoamnios asociaban los siguientes diagnósticos: malformación renal o de la vía urinaria (6 casos), rotura prematura de membranas (5 casos, 3 de estos debido a intervención prenatal invasiva), hidropesía fetal (3 casos), hernia congénita diafragmática (un caso) y en uno no se estableció agente causal. Los casos de hernia diafragmática congénita mostraron una LHR media de 0,89 (0,29-4,6). Quince pacientes fueron sometidos a intervención quirúrgica fetal el tras hallazgo de alteración ecográfica, 11 casos sometidos a FETO y 5 casos con drenajes toracoamnióticos.
Distribución causal y evolución de los casos diagnosticados de hipoplasia pulmonar entre 1995 y 2014
| Causas de hipoplasia pulmonar | Número de casos (%) | Pacientes fallecidos | Pacientes que requirieron ventilación mecánica en el periodo neonatal | Pacientes con ventilación mecánica domiciliaria |
|---|---|---|---|---|
| Hernia diafragmática | 24 (40) | 9 | 23 | 1 |
| Izquierda | 16 | |||
| Bilateral | 3 | |||
| Derecha | 5 | |||
| Eventración-parálisis diafragmática | 2 (3,3) | 1 | 1 | |
| Malformación renal/vía urinaria | 6 (10) | 2 | 6 | 3 |
| Intervencionismo prenatal invasivo | 3 (5) | 3 | 3 | |
| Hidropesía fetal | 3 (5) | 3 | 3 | |
| Rotura prematura de membranas | 2 (3,3) | 2 | 2 | |
| Mecanismo oligoamnios no causa aclarada | 2 (3,3) | 1 | ||
| Hidrotórax congénito | 2 (3,3) | 1 | 1 | |
| Displasia torácica | 2 (3,3) | 1 | 1 | 1 |
| Síndrome de cimitarra | 5 (8,3) | |||
| Masas intratorácicas | 2 (3,3) | 1 | 1 | |
| Etiología no aclarada (asociado a síndrome de Down) | 1 (1,6) | 1 | 1 | |
| Secuencia acinesia/hipocinesia fetal | 2 (3,3) | 2 | 2 | |
| Primaria | 4 (6,6) | 2 | 1 | 1 |
La media de edad gestacional al nacer fue de 33 semanas (27-39 semanas). Cincuenta casos presentaron dificultad respiratoria al nacer y un 94% (47 casos) de estos requirieron ventilación mecánica por hipoxemia grave, durante 9 días de media. Ocho pacientes presentaron hipertensión pulmonar persistente del recién nacido.
En 6 pacientes la sintomatología fue más tardía, siendo diagnosticados de HP tras ser estudiados por episodios de bronquitis recidivantes. La media de edad al diagnóstico de estos 6 casos fue de 45 días de vida (30-180). Cuatro casos no mostraron ninguna sintomatología clínica a nivel respiratorio y el diagnóstico de HP se hizo tras el hallazgo de dextrocardia en el primer año de vida, con posterior confirmación de síndrome de cimitarra en todos ellos.
El diagnóstico de sospecha de HP se estableció en 18 pacientes (31%) con muerte neonatal temprana, mediante criterios clínicos y radiografía simple de tórax compatible. En 31 casos (51,6%) el diagnóstico se estableció mediante TC torácica o gammagrafía pulmonar de perfusión: 14 casos con las 2 técnicas, 11 mediante TC y 6 mediante gammagrafía únicamente. En los casos en que se utilizó gammagrafía el valor promedio de perfusión del pulmón afectado fue del 29% (8-44%). En 11 pacientes (19%) el diagnóstico se confirmó mediante necropsia.
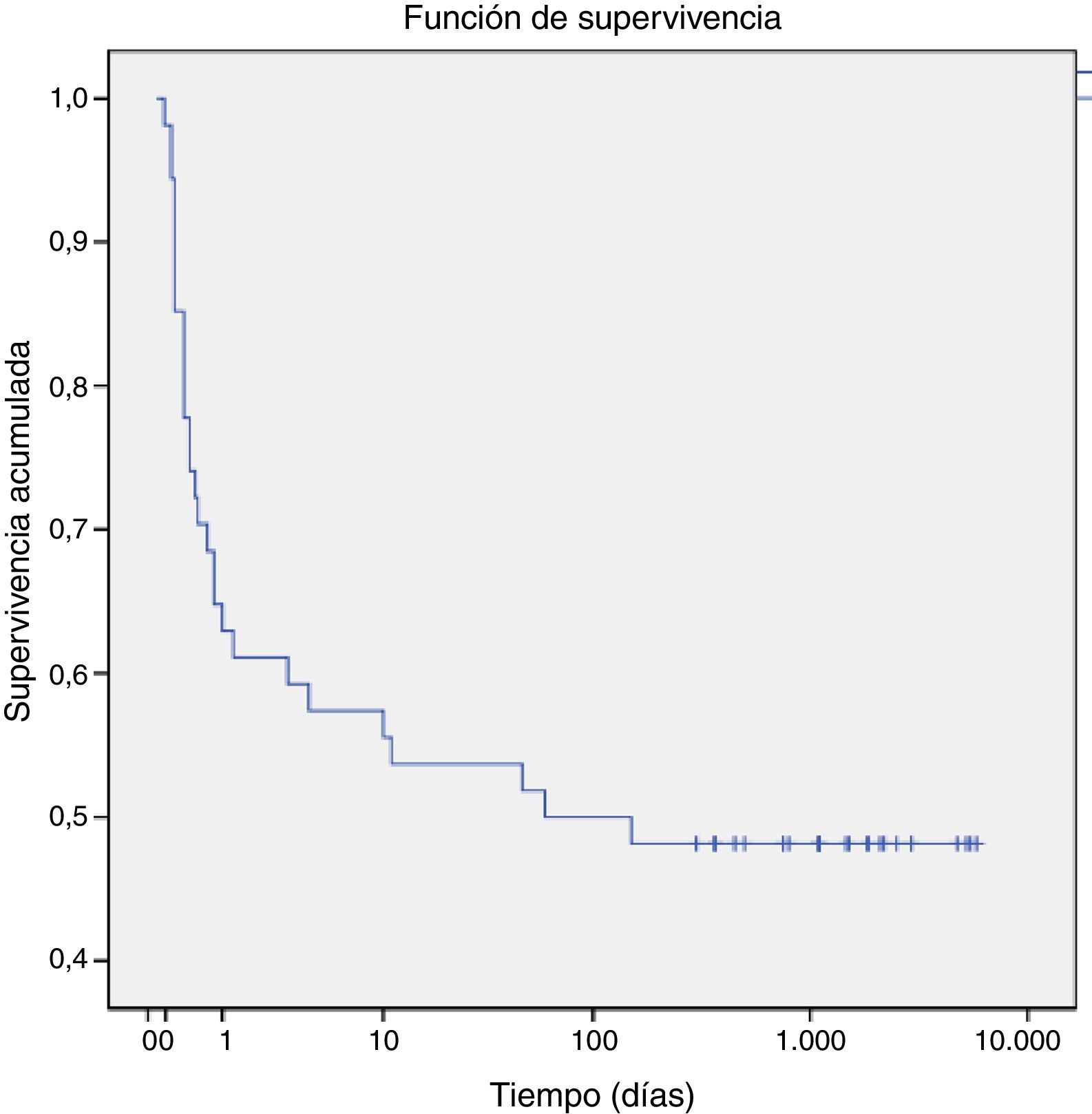
Respecto a la mortalidad, un total de 28 pacientes (47%) fallecieron antes de los 60 días de vida, 25 murieron en el periodo perinatal. El 75% de los fallecimientos fue en el primer día de vida. El análisis de supervivencia de Kaplan-Meier (fig. 1) muestra una probabilidad acumulada de muerte del 25% antes de las 12 primeras horas de vida, que aumentó al 50% a los 60 días de vida.
Tras 4 pérdidas de seguimiento, se ha realizado seguimiento a 28 pacientes durante una media de 5,8 años (0,9-16). De los recién nacidos que requirieron soporte ventilatorio inicialmente, 6 pacientes requirieron posteriormente ventilación mecánica domiciliaria (2 mediante traqueostomía) durante una mediana de 36 meses y 5 pacientes requirieron oxigenoterapia domiciliaria durante una mediana de 4 meses (rango 2-10 meses). Un paciente desarrolló hipertensión pulmonar. Diecisiete presentaron clínica respiratoria recidivante catalogada de neumonía y bronquitis de repetición, y 4 desarrollaron escoliosis. Se analizaron las pruebas de función pulmonar de los últimos 5 años (tabla 3). La espirometría fue técnicamente aceptable en 13/28 (42,8%) pacientes (rango edad 5-17 años); de estos, 13 niños, 11 tenían pletismografía técnicamente correcta. Cinco pacientes presentaron patrón obstructivo (FEV1/FVC) < LLN (del inglés lower limits of normal), 2 presentaron patrón restrictivo (FVC<límite inferior normal LN y FEV1/FVC>LLN y TLC>LLN), y 6 patrón normal, donde 2 mostraron atrapamiento aéreo. Tres mostraron resistencias aumentadas. Los estudios de difusión en los 11 pacientes fueron normales.
Resultados de las pruebas de función pulmonar estudiadas en los últimos 5 años en niños afectados de hipoplasia pulmonar diagnosticados entre 1995 y 2014 en el Hospital Universitario Vall d’Hebron de Barcelona
| Número de paciente | Z-score FVCa | Z-score FEV1a | Z-score FEV1/FVC | Z-score TLCb | Interpretación |
|---|---|---|---|---|---|
| 1 | –2,06 | –2,7 | –1,55 | – | Obstructivo |
| 2 | –2,85 | –3,36 | –1,70 | –0,59 | Obstructivo |
| 3 | 1,18 | –0,37 | –2,13 | – | Normal |
| 4 | –2,29 | –2,41 | –0,44 | –1,88 | Restrictivo |
| 5 | –2,67 | –3,43 | –1,90 | –0,69 | Obstructivo |
| 6 | –0,19 | 0,08 | 0,37 | 0,05 | Normal |
| 7 | –2,12 | –3,92 | –3,12 | –0,25 | Obstructivo |
| 8 | –2,84 | –2,7 | 0,15 | –2,72 | Restrictivo |
| 9 | –0,49 | –1,25 | –1,35 | 1,96 | Normal |
| 10 | –0,9 | –1,01 | –0,37 | –0,08 | Normal |
| 11 | 0,67 | –1,14 | –1,00 | 0,14 | Normal |
| 12 | –2,48 | –2,24 | 0,35 | –0,31 | Obstructivo |
| 13 | 0,73 | 0,84 | 0,00 | 0,42 | Normal |
| Mediana Z-score (rango) | –2,06 (–2,84 a 1,18) | –2,55 (–3,92 a 0,84) | –1 (–3,12 a 0,37) | –0,25 (–1,88 a 1,96) |
Veintidós pacientes pudieron realizar una escolarización normal y describieron una correcta tolerancia al ejercicio para su edad.
DiscusiónLa HP es el resultado de la interrupción del desarrollo pulmonar normal debido a distintos factores aún no totalmente conocidos. Se presenta una revisión detallada de los casos diagnosticados en los últimos 20 años, atendidos por un equipo multidisciplinar de un hospital universitario de alto nivel de complejidad. Se analizan sus múltiples causas, presentación clínica, técnicas diagnósticas, tratamientos y evolución.
La distribución etiológica de los casos descritos en nuestro trabajo coincide con los datos de la literatura8,20,21, donde las principales alteraciones congénitas que se asocian a HP son: hernia diafragmática, anomalías renales y anomalías esqueléticas. Cabe destacar otras entidades encontradas en nuestra revisión como rotura prematura de membranas por técnicas intervencionistas prenatales, tales como fetorredución o amniocentesis, alteraciones genéticas (trisomía 21), y algunas menos conocidas, como la secuencia de acinesia/hipocinesia fetal o síndrome de Pena-Shokeir (2 casos en nuestra serie), que se caracteriza por polihidramnios, retraso de crecimiento intrauterino, anomalías craneofaciales y de extremidades, contracturas musculares, cordón umbilical corto y HP, con supervivencia muy limitada22,23. Los casos de HP primaria son infrecuentes, pudiendo ser unilaterales o lobares3. En nuestra serie, únicamente el 7,8% de los casos fue catalogado de HP primaria.
En relación con la presencia de oligoamnios, aproximadamente un tercio de los nuestros casos asociaron este mecanismo de HP. Nimrod et al.18 describían un mayor impacto de la rotura prematura de membranas cuando ocurre en fetos menores de 26 semanas y con un oligoamnios superior a 5 semanas; Rotschild et al.20 detectaron hipoplasia pulmonar hasta en un 14% de fetos menores de 29 semanas expuestos a 7 días de bajo nivel de líquido amniótico tras la rotura.
Otros autores han establecido una correlación entre el riesgo de presentar HP con el oligoamnios y la edad gestacional, concluyendo que la presencia de oligoamnios durante más de 14 días tras una rotura en menores de 25 semanas de edad gestacional tiene un riesgo de mortalidad por HP de hasta el 90%21,22. Aunque en nuestra serie no hemos establecido subgrupos para el estudio de oligoamnios, en cuanto a su duración y la edad gestacional en el momento del diagnóstico, hemos observado que la edad media al hallazgo de oligoamnios era de 27 semanas de gestación, hecho que coincidiría con los datos publicados.
Paralelamente a la literatura, la malformación congénita más frecuentemente relacionada con HP fue la hernia diafragmática10,24,25. Esta entidad es el modelo que más se ha utilizado para el estudio de intervenciones precoces, de la fisiopatología, avances en cuidados perinatales y desarrollo de nuevas técnicas diagnósticas de HP26. La hernia congénita diafragmática tiene la particularidad de que el desarrollo de HP es inherente a la misma patología y no solamente debido al efecto compresivo, siendo todavía desconocido el mecanismo por el cual también se afecta el pulmón contralateral al defecto herniario17,18.
El diagnóstico prenatal de HP y su confirmación en la etapa posnatal precoz es de gran importancia para disminuir la morbimortalidad de esta patología. Por este motivo, se ha estandarizado el uso de ecografía tridimensional y RM para el diagnóstico prenatal13,16,27,28. Actualmente, las recomendaciones de la ERS sobre el manejo de la hernia diafragmática congénita incluyen la RM en el estudio prenatal de los afectados, por su superior capacidad respecto a la ecografía para identificar la herniación hepática al tórax, diferenciar una eventración diafragmática y realizar una medición más certera del volumen pulmonar contralateral e ipsolateral al defecto herniario29. Levine et al.27 realizaron RM a 74 fetos diagnosticados de anomalías torácicas tras ecografía, confirmando que el uso de otra técnica complementaria no invasiva aportaba información adicional hasta en un 38% de los casos y modificaba el tratamiento en el 8% de los mismos. Todos nuestros pacientes fueron estudiados mediante ecografía tridimensional durante el periodo prenatal y a los casos graves de hernia diafragmática también se les realizó RM.
Entre los 24 pacientes diagnosticados de hernia diafragmática congénita como causa de HP, 11 fueron clasificados como casos de riesgo de alta mortalidad cumpliendo criterios para intervención prenatal quirúrgica, por lo que fueron sometidos a técnica FETO. Las técnicas invasivas de intervencionismo prenatal han evolucionado desde el pinzamiento abierto de la tráquea fetal, la implantación de un tapón traqueal, hasta la técnica FETO en la cual, tras comprobar el crecimiento pulmonar, se retira el globo prenatalmente. Estas técnicas han conllevado un aumento de la supervivencia posnatal y una disminución de la hipertensión pulmonar secundaria15,30,31.
Tras el nacimiento, el diagnóstico puede sospecharse mediante radiología simple, cuando observamos cúpulas diafragmáticas por encima de la séptima costilla, tórax acampanado, costillas inclinadas hacia abajo, neumotórax o neumomediastino2,3,32. Las técnicas de gammagrafía pulmonar de perfusión y TC torácica son muy útiles para la confirmación diagnóstica5,32-37. En nuestra serie, el 50,4% de los casos fueron diagnosticados mediante las técnicas de gammagrafía y TC; el 31% de los casos con radiología simple y clínica compatible (los cuales no pudieron ser confirmados por otras técnicas porque fallecieron de forma precoz), y el 19% mediante necropsia. El valor promedio de perfusión en la gammagrafía hallado corrobora la gravedad de los casos, catalogándolos de afectación moderada cuando la perfusión del pulmón afectado es inferior al 40%33-35.
La tasa de mortalidad perinatal observada en nuestro grupo fue del 41,6%, coincidiendo con las distintas series publicadas que describen una elevada mortalidad perinatal2,4,10,15,25. El análisis de nuestros datos muestra que la probabilidad acumulada de muerte fue del 25% antes de las 12 primeras horas de vida y que a los 60 días de vida esta aumentaba hasta el 50%, hecho que confirma la necesidad de disponer de un equipo multidisciplinar altamente especializado para el manejo de estos pacientes durante la etapa neonatal. A lo largo del seguimiento, las manifestaciones clínicas más frecuentes observadas fueron neumonías y bronquitis recidivantes, coincidiendo con los evoluciones descritas de pacientes con hernia diafragmática congénita corregida y otros casos de diagnóstico tardío que evidencian síntomas similares29,37. Un 25% de los pacientes que requirieron ventilación mecánica inicialmente, y sobrevivieron, precisaron ventilación mecánica domiciliaria y dependencia de oxígeno durante una mediana de 4 meses. Estos datos contrastan con la literatura, donde el tiempo de soporte respiratorio es menor34. Ello puede ser debido a que no existen estudios descriptivos similares al de nuestra población. Por otro lado, un 16% de nuestros pacientes desarrolló escoliosis asociada a la disminución del volumen pulmonar unilateral. Estudios han observado que hasta un 16% de niños con diagnóstico previo de hernia diafragmática presentan alguna asimetría torácica, escoliosis o pectum carinatum34. Otros estudios de seguimiento en niños con hernia diafragmática corregida muestran un crecimiento recuperador del tamaño pulmonar, con buena tolerancia al ejercicio, pudiendo realizar vida normal, a pesar de que la asimetría en las imágenes radiológicas persiste y los estudios de perfusión se mantienen sin cambios19. De forma similar, en nuestra serie un 77% de los pacientes supervivientes pudieron realizar una escolarización normal y presentaron una correcta tolerancia al ejercicio para su edad.
Respecto a los estudios de función pulmonar, algunos autores describen un patrón obstructivo en pacientes con HP y proponen la necesidad de realizar broncoscopia para descartar traqueobroncomalacia38. La mayoría de las publicaciones muestran un patrón restrictivo predominante, sobre todo en pacientes que requirieron ventilación mecánica u oxigenoterapia de forma prolongada39,40. En nuestra serie, disponemos de escasos datos de función pulmonar, debido a que solo se han considerado en esta publicación los recogidos durante los últimos 5 años porque fueron realizados por los mismos técnicos, con el mismo equipo y estrictos criterios de calidad y aceptabilidad publicados por la ERS para evitar sesgos. Además, la mayor parte de pacientes en seguimiento son todavía no colaboradores para espirometría o pletismografía. Nuestros resultados actuales muestran una mayor proporción de niños con patrón obstructivo sobre el restrictivo en el seguimiento, tanto en la espirometría como en la pletismografía.
La HP es una entidad donde en las últimas 2 décadas han evolucionado las técnicas diagnósticas e intervenciones prenatales y se han optimizado los cuidados posnatales. Esto ha permitido mejorar la asistencia a estos pacientes, aunque la morbimortalidad asociada sigue siendo importante. Creemos que aún faltan estudios que permitan medir el valor de estas intervenciones precoces sobre el posterior desarrollo pulmonar. Así mismo, es necesario continuar investigando para poder identificar nuevos factores que permitan modificar el curso de la HP de forma temprana y menos invasiva.
ConclusionesLa HP es una malformación congénita de etiología diversa, con una frecuencia subestimada. Presenta una elevada mortalidad neonatal y una importante morbilidad a largo plazo, por lo que requiere de un diagnóstico temprano y la intervención de un equipo multidisciplinar especializado. El desarrollo de nuevas técnicas diagnósticas y quirúrgicas ha permitido mejorar la supervivencia de estos pacientes, aunque sigue siendo necesario seguir investigando para poder identificar nuevos factores que permitan modificar la historia natural de esta entidad y estudios a largo plazo que evalúen el efecto de dichas intervenciones.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
