









La hipofosfatasia es una enfermedad ultra-rara del metabolismo mineral óseo causada por un déficit de actividad de la fosfatasa alcalina, debido a la existencia de mutaciones en el gen ALPL. Clínicamente, se caracteriza por el desarrollo de hipomineralización esquelética y dental, junto con la frecuente aparición de manifestaciones extraesqueléticas. Su espectro fenotípico es muy variable y engloba una forma de afectación exclusivamente odontológica (odontohipofosfatasia) y 5 subtipos de afectación sistémica diferenciados según el momento de inicio de los síntomas (4 de los cuales se desarrollan en la edad pediátrica: formas perinatal letal, perinatal benigna, del lactante e infanto-juvenil). Las formas de inicio más precoz presentan, generalmente, peor pronóstico, debido a la posibilidad de desarrollar complicaciones potencialmente letales, como la dificultad respiratoria grave por malformaciones torácicas o la presencia de convulsiones. Debido a la baja prevalencia de las formas graves de la enfermedad, y a su variabilidad y solapamiento fenotípico con otras patologías más prevalentes, el diagnóstico de la hipofosfastasia en la práctica clínica constituye un reto. No obstante, su potencial gravedad e impacto sobre la calidad de vida de los pacientes, así como la reciente disponibilidad de un tratamiento de reemplazo enzimático específico, confieren particular relevancia a la correcta identificación de los pacientes afectos de hipofosfatasia. A partir de la evidencia publicada y la experiencia clínica, en el presente artículo se propone un algoritmo con recomendaciones prácticas para el diagnóstico diferencial de la hipofosfatasia en niños, así como una revisión actualizada de las opciones de tratamiento existentes.
Hypophosphatasia is a very rare bone metabolism disorder caused by a deficiency in alkaline phosphatase activity, due to mutations in the ALPL gene. Its clinical hallmark is the impairment of skeletal and teeth mineralisation, although extra-skeletal manifestations are frequent. Its phenotypic spectrum is widely variable from a subtype with exclusive odontological impairment (odontohypophosphatasia) to five subtypes with systemic involvement, classified according to the age at the onset of the first symptoms (four of them in the paediatric age range: perinatal lethal, perinatal benign, infant and childhood hypophosphatasia). Those subtypes of hypophosphatasia with an earliest onset usually involve a worse prognosis, due to the risk of developing potentially lethal complications, such as seizures or severe respiratory insufficiency, secondary to rib cage malformations.
Due to the extremely low prevalence of the severe forms of hypophosphatasia, its clinical variability and overlapping phenotypic features with several more prevalent conditions, the diagnosis of hypophosphatasia in the clinical setting is challenging. However, its potential lethality and impact on the patient's quality of life, along with the recent availability of an enzyme replacement therapy, increases the relevance of the early and accurate identification of patients affected with hypophosphatasia.
On the basis of published evidence and clinical experience, this article suggests an algorithm with practical recommendations for the differential diagnosis of childhood hypophosphatasia, as well as an updated review of current therapeutic options.
La hipofosfatasia (HPP) es una enfermedad «ultra-rara» hereditaria del metabolismo calcio-fósforo, progresiva y sistémica, caracterizada por una hipomineralización esquelética secundaria a una actividad deficiente de la fosfatasa alcalina (FA) no específica de tejido (TNSALP)1,2. La FA es una enzima esencial para el proceso de mineralización ósea y dental, existiendo al menos 4 isoenzimas: TNSALP (95% de la actividad sérica total de la FA) y 3 isoenzimas específicas de tejido (intestinal, placentaria y de células germinales)1,2. TNSALP está codificada por el gen ALPL (1p36.12; MIM 171760), expresado principalmente en hígado, hueso, riñón, músculo y en áreas sensoriales del córtex cerebral3. La presencia de variantes que afectan a la función de ALPL determinan el desarrollo de HPP, habiéndose descrito más de 330 mutaciones4.
Los principales sustratos de TNSALP son el pirofosfato inorgánico (PPi), el piridoxal 5′-fosfato (PLP) y la fosfoetanolamina (PEA)3. En pacientes con HPP, la actividad deficiente de TNSALP determina su acúmulo y el desarrollo de manifestaciones de la enfermedad. La elevada concentración de PPi en la matriz ósea extracelular inhibe la nucleación de cristales de calcio y fosfato, interfiriendo en la formación de cristales de hidroxiapatita e induciendo alteraciones esqueléticas3. Por su parte, la desfosforilación del PLP (metabolito activo de la vitamina B6 o piridoxina) a piridoxal (PL), mediante la acción de TNSALP, es necesaria para que la piridoxina pueda atravesar la membrana celular o la barrera hematoencefálica. El PLP es un cofactor esencial de neurotransmisores inhibidores como el ácido gamma-amino-butírico. En las formas clínicas neonatales y del lactante de la HPP son características las convulsiones influidas por la deficiencia central de vitamina B6, pese al acúmulo plasmático de PLP1–3. Adicionalmente, la presencia frecuente de daño inflamatorio articular en los pacientes con HPP indica una implicación de TNSALP en procesos inflamatorios activados por depósitos de dihidrato de pirofosfato cálcico.
La HPP se clasifica tradicionalmente en una forma de afectación exclusivamente odontológica (odontohipofosfatasia) y en 5 subtipos de afectación sistémica según el momento de inicio de los síntomas (4 formas pediátricas y una adulta) (tablas 1–3). Actualmente, es necesario diferenciar, al menos en el subtipo infanto-juvenil, entre formas graves y leves1. Además, se ha descrito otra entidad con clínica similar a la HPP del lactante pero con una actividad de FA sérica normal o incluso ligeramente aumentada (seudohipofosfatasia)1, aunque podría deberse circunstancias intercurrentes1.
Características clínicas de las formas perinatales de hipofosfatasia
| Subtipos | Edad de inicio sintomatología | Principales manifestaciones clínicas | Pruebas diagnósticas | Evolución natural |
|---|---|---|---|---|
| Forma perinatal letal (herencia AR) | Periodo perinatal (signos ecográficos observables ya intraútero) | Alteraciones esqueléticas12,20 Hipomineralización grave Acortamiento e incurvación de los huesos largos Ausencia de osificación de la bóveda craneal, a excepción de la calcificación central (caput membranaceum) Espolones osteocondrales en ulnas y fíbulas (característicos) Fracturas y fragilidad ósea Alteraciones neurológicas Convulsiones dependientes de vitamina B6 Hipersensibilidad a los estímulos (irritabilidad) Hemorragia intracraneal (posible) Otras alteraciones21 Hipoplasia pulmonar grave (secundaria a las malformaciones torácicas) Apnea patológica Fiebre idiopática Anemia mieloptísica Hipercalcemia Hiperfosfatemia | Pruebas de imagen Radiografía: alteraciones esqueléticas típicas asociadas Ecografía: malformaciones típicas asociadas Pruebas de laboratorio Actividad FA total (en sangre de cordón umbilical): baja Análisis genético/diagnóstico prenatal (en sangre de cordón umbilical o biopsia de vellosidades coriónicas) Mutaciones patogénicas en el gen ALPL | Forma más grave, supervivencia a largo plazo mínima La muerte fetal intrauterina o muerte neonatal en los primeros días/semanas son frecuentes |
| Forma perinatal benigna (herencia AR/AD) | Periodo perinatal (signos ecográficos observables ya intraútero) | Alteraciones esqueléticas Hipomineralización Acortamiento e incurvación de huesos largos | Pruebas de imagen Radiografía: alteraciones esqueléticas típicas asociadas Ecografía: malformaciones típicas asociadas Pruebas de laboratorio Actividad FA total (en sangre de cordón umbilical): baja Análisis genético/diagnóstico prenatal Mutación patogénica en el gen ALPL | Mejora espontánea de las alteraciones esqueléticas tras el parto con un pronóstico favorable (se desconoce la evolución natural a largo plazo) |
AD: autosómica dominante; ALPL: Gen de la fosfatasa alcalina no específica de tejido; AR: autosómica recesiva; FA: fosfatasa alcalina.
Características clínicas de la hipofosfatasia del lactante y la odontohipofosfatasia
| Subtipos | Edad de inicio sintomatología | Principales manifestaciones clínicas | Pruebas diagnósticas | Evolución natural |
|---|---|---|---|---|
| Forma del lactante (herencia AR) | Antes de los 6 meses de vida | Alteraciones esqueléticas Hipomineralización grave Tórax batiente y/o con deformidad raquítica Fracturas y fragilidad ósea Fallo de medro Alteraciones craneales y faciales5 Craneosinostosis Malformación de Arnold-Chiari tipo i Hidrocefalia hidrostática Hidrosiringomielia Aumento de la presión intracraneal con papiledema Proptosis. Hipertelorismo leve Dolicocefalia. Escleróticas azules Alteraciones neurológicas13 Convulsiones sensibles a vitamina B6 Hipersensibilidad a los estímulos (irritabilidad) Otras alteraciones Trastornos de la deglución Hipotonía grave. Debilidad/escasa fuerza muscular Hipercalcemia (anorexia, vómitos). Hipercalciuria/nefrocalcinosis Disfunción/compromiso renal | Anamnesis completa Pruebas de imagen Radiografía: alteraciones esqueléticas típicas asociadas Densitometría ósea: baja densidad ósea Pruebas de laboratorio Actividad FA total: baja Sustratos de la FA elevados: PLP en plasma, PEA orina Análisis genético (en sangre): Mutaciones patogénicas en el gen ALPL | Forma con pronóstico variable en función de la gravedad de las manifestaciones clínicas Los pacientes con convulsiones presentan un peor pronóstico5,11,13 |
| Odontohipofosfatasia (herencia AR/AD) | A cualquier edad tras el inicio de la dentición | Alteraciones dentales Pérdida prematura de la dentición decidua y/o la permanente Caries dental grave. Grosor de la dentina reducido. Agrandamiento de las cámaras pulpares Pérdida del hueso alveolar Otras alteraciones No asociada a alteraciones óseas, articulares o musculares | Evaluación clínica y dental Pruebas de laboratorio Actividad FA total: baja Sustratos de la FA elevados: PLP en plasma, PEA en plasma y orina Análisis genético (en sangre): Mutación en el gen ALPL | Forma con manifestaciones clínicas menos graves y mejor pronóstico |
AD: autosómica dominante; ALPL: gen de la fosfatasa alcalina no específica de tejido; AR: autosómica recesiva; FA: fosfatasa alcalina; PEA: fosfoetanolamina; PLP: piridoxal 5′-fosfato.
Características clínicas de la hipofosfatasia infanto-juvenil
| Subtipos | Edad de inicio sintomatología | Principales manifestaciones clínicas | Pruebas diagnósticas | Evolución natural |
|---|---|---|---|---|
| Infanto-juvenil (herencia AR/AD) | Después de los 6 meses de vida | Alteraciones esqueléticas12,22 Hipomineralización/deformidades raquíticas Fallo de medro, talla baja Fracturas recurrentes Acortamiento e incurvación de huesos largos «rosario costal»/deformidad raquítica costal Ensanchamiento metafisario Escoliosis Densidad mineral ósea reducida Alteraciones craneales y faciales22: Craneosinostosis Aumento de la presión intracraneal Proptosis Dolicocefalia Alteraciones psicomotrices23 Retraso en la adquisición de hitos del desarrollo motor (deambulación libre) Marcha anadeante (de pato) Alteraciones dentales5,11,16 Retraso en la erupción y pérdida prematura de la dentición, decidua sin reabsorción de las raíces (característico) Agrandamiento de las cámaras pulpares Pérdida del hueso alveolar Caries frecuentes y graves Otras alteraciones23 Dolor óseo y articular crónico Hipotonía muscular Fatigabilidad exagerada Falta de apetito Trastornos gastrointestinales | Anamnesis completa. Pruebas de imagen Radiografía: alteraciones esqueléticas típicas asociadas Densitometría mineral ósea: baja densidad ósea Pruebas de laboratorio Actividad FA total: baja Sustratos de la FA elevados: PLP en plasma, PEA en plasma y orina Análisis genético (en sangre): mutación patogénica en el gen ALPL | Forma con pronóstico variable en función de la gravedad de las manifestaciones clínicas En ocasiones las alteraciones óseas presentan remisión espontánea tras la pubertad, pudiendo reaparecer en la edad adulta5,11 |
AD: autosómica dominante; ALPL: Gen de la fosfatasa alcalina no específica de tejido; AR: autosómica recesiva; FA: fosfatasa alcalina; PEA: fosfoetanolamina; PLP: piridoxal 5′-fosfato.
Generalmente, las formas de inicio más temprano presentan peor pronóstico1,2,5 debido a las complicaciones potencialmente letales de sus alteraciones esqueléticas (dificultad respiratoria grave por malformaciones torácicas) y extraesqueléticas (presencia de convulsiones)6.
Las formas graves de HPP tienen una prevalencia estimada de 1 caso/300.000 nacimientos en Europa5. La frecuencia varía entre poblaciones, siendo las formas graves más frecuentes en los menonitas canadienses (1:2.500)2 y las formas perinatales letales en Japón7. Las formas leves, de herencia dominante, tienen una prevalencia estimada de 1/6.370 en Europa8. En contraste, la prevalencia de hipofosfatasemia (niveles bajos de actividad de FA sérica) es muy superior, siendo múltiples las causas que, en ausencia de HPP, pueden determinarla (tabla 4)1.
Situaciones que pueden presentar niveles bajos de actividad de la fosfatasa alcalina
| Niveles bajos de actividad de la fosfatasa alcalina | |
|---|---|
| Patologías | Situaciones clínicas/metodológicas de laboratorio |
| Anemia perniciosa o intensa Celiaquía Deficiencia de vitamina C Deficiencia de cinc o magnesio Displasia cleidocraneal Enfermedad de Wilson Hipotiroidismo Intoxicación por vitamina D Metales pesados radiactivos Mieloma múltiple Osteogénesis imperfecta tipo ii Síndrome lácteo-alcalino Síndrome de Cushing | Inmovilización prolongada Cirugía de derivación cardíaca (bypass) Desnutrición Inanición Muestra de sangre recogida de forma inadecuada (oxalato, EDTA) Rangos de referencia inapropiados Terapia con clofibrato Transfusión de sangre o plasma |
EDTA: ácido etildiaminotetraacético.
El diagnóstico de HPP en la práctica clínica es un reto por su baja prevalencia y el gran solapamiento fenotípico con otras patologías más prevalentes. Además, el escaso valor conferido habitualmente a una actividad sérica de FA disminuida (en contraste con la valoración que habitualmente se realiza de la hiperfosfatasemia) es un factor limitante para el diagnóstico. No obstante, el diagnóstico es particularmente relevante por la potencial gravedad de la enfermedad, su intensa afectación de la calidad de vida en muchos pacientes, o la posible iatrogenia derivada de un diagnóstico equívoco. Además, la reciente disponibilidad de un tratamiento de reemplazo enzimático específico de la HPP9,10 determina la necesidad de un diagnóstico correcto en aras de un tratamiento precoz y adecuado.
El principal objetivo de este artículo es proporcionar recomendaciones generales para facilitar la identificación de pacientes pediátricos afectados de HPP. Para un conocimiento detallado de las bases genéticas, fisiopatológicas y clínicas de la enfermedad, se recomienda la consulta de revisiones recientes de interés1,2,6,10.
Diagnóstico de la hipofosfatasiaLa figura 1 muestra un algoritmo para el diagnóstico de la HPP en niños, basada en la evidencia publicada y en recomendaciones de expertos derivadas de la experiencia clínica acumulada. Se pretende establecer una secuencia diagnóstica razonada ante potenciales casos de HPP clínicamente expresiva, aunque existen formas con mínima o nula expresividad clínica en el periodo infanto-juvenil. En estos casos, el hallazgo de niveles persistentemente bajos de actividad de FA sérica, aún en ausencia de manifestaciones esqueléticas, puede indicar la existencia de una HPP paucisintomática (que puede tener mayor expresividad clínica en la edad adulta). El diagnóstico de HPP debe considerarse fundamentalmente en pacientes que presentan ciertos signos y/o síntomas músculo-esqueléticos y dentales (aunque en las formas graves perinatales y del lactante los síntomas respiratorios y neurológicos apoyan fuertemente la sospecha diagnóstica). La tablas 1–3 resumen las manifestaciones clínicas más habituales de las formas pediátricas de HPP.
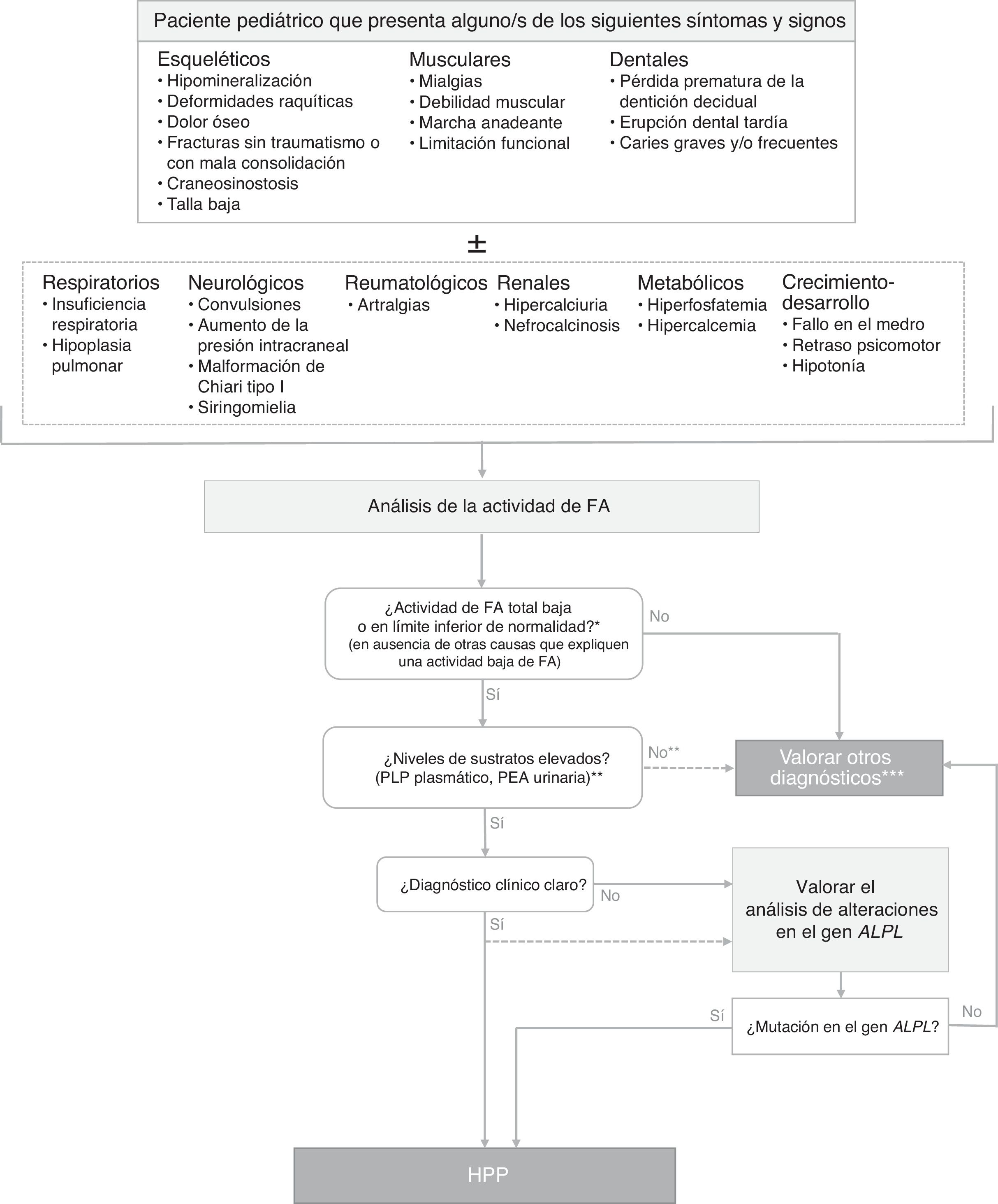
Algoritmo de diagnóstico de la hipofosfatasia en pacientes pediátricos.
ALPL: gen de la fosfatasa alcalina no específica de tejido; FA: fosfatasa alcalina; HPP: hipofosfatasia; PEA: fosfoetanolamina; PLP: piridoxal 5′-fosfato.
* En ≥ 2 determinaciones, sin antecedentes de FA normal y fuera de una situación aguda.
** El PEA es un parámetro poco específico, no siempre está elevado en HPP. Los pacientes con HPP tienen a menudo una actividad total de la FA en el límite de detección, mientras que en las formas menos severas la actividad de FA puede estar en el límite de la normalidad o algo disminuidas. La reducción de la actividad de FA da como resultado la acumulación de sustratos.
*** En estos pacientes en los que no es posible hallar otras causas que expliquen el cuadro clínico, no debe descartarse el diagnóstico de HPP, independientemente de los valores de los sustratos de la FA.
En la forma perinatal letal los hallazgos radiológicos son altamente indicativos de la enfermedad: ausencia de mineralización de algunos huesos (constituyendo la osteogénesis imperfecta su principal diagnóstico diferencial), caput membranaceum y prominencias osteocondrales («espolones») en antebrazos y piernas (rasgo patognomónico) (fig. 2 a)2. A diferencia de la hipocalcemia e hipofosfatemia observada en distintos tipos de raquitismo, la HPP puede cursar con hipercalcemia e hiperfosfatemia (50%), siendo excepcional la presencia de niveles séricos bajos de calcio y fósforo en esta enfermedad11. La causa de la hipercalcemia es desconocida, aunque podría deberse a la falta de incorporación ósea de calcio. La hiperfosfatemia es debida a un incremento en la reabsorción tubular de fosfato1.
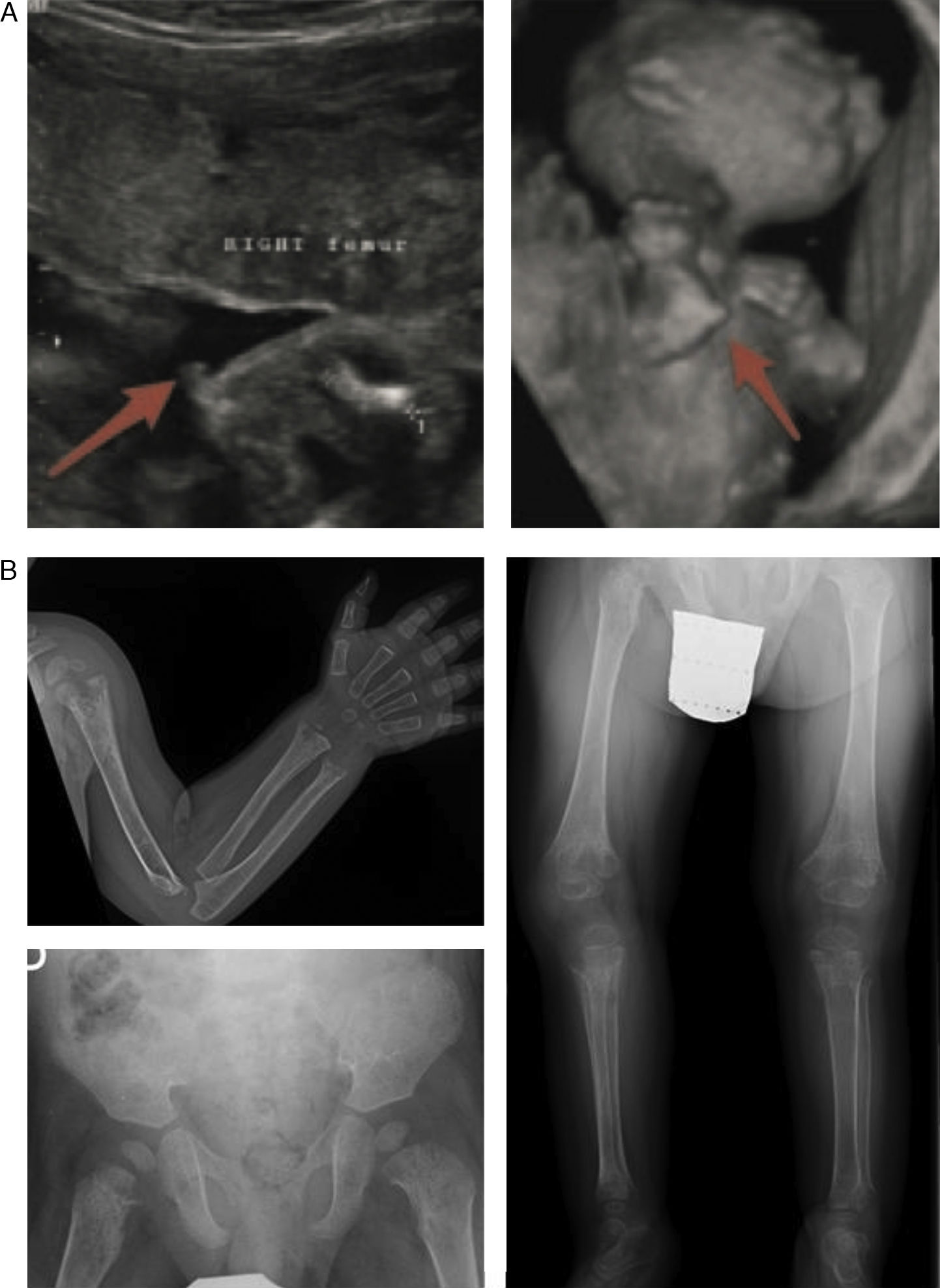
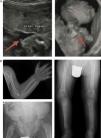
Hallazgos característicos en las pruebas de imagen de la hipofosfatasia perinatal letal (a) y de la hipofosfatasia del lactante (b). a) Ultrasonografía a las 18 semanas de gestación: espolones óseos en la rodilla derecha y en el codo derecho (3D). b) Estudio radiológico en paciente de 17 meses de edad: marcada alteración en metáfisis de huesos largos (proximales de ambos húmeros, distales de ambos radios y cúbitos y proximales de ambos fémures, tibias y peronés), que se encuentran ensanchadas, con pérdida de densidad ósea, trabéculas groseras y proyecciones radiolucentes que se extienden desde la fisis hacia la metáfisis. Reacción perióstica lineal en el radio izquierdo.
Imágenes reproducidas con permiso de Zankl A, Mornet E, Wong S. Specific ultrasonography features of perinatal lethal hypophosphatasia. Am J Med Gen Part A. 2008;146A:1200-1204, y Caballero Mora FJ, Martos Moreno GA, García Esparza E, Argente J. Hipofosfatasia infantil. An Pediatr. 2012;76:368-369.
En la forma perinatal benigna las ecografías gestacionales muestran signos de hipomineralización en los huesos de las extremidades, aunque a partir del tercer trimestre de embarazo los pacientes mejoran espontáneamente y pueden tener un desarrollo normal.
Los signos más característicos de la HPP del lactante son los dolores osteomusculares (con/sin fracturas), deformidades esqueléticas similares a las observadas en el raquitismo, fallo de medro, hipotonía/debilidad muscular y retraso en la adquisición de los hitos del desarrollo psicomotor6. Las alteraciones esqueléticas más habituales son la deformidad raquítica costal, incurvación de los huesos largos, genu varum (tras iniciar bipedestación), fracturas y craneosinostosis. Esto se corresponde con la presencia en las pruebas de imagen de ensanchamiento, desflecamiento y áreas de radiolucencia en las metáfisis de los huesos largos5,11,12 (fig. 2 b).
El fenotipo de las formas más graves de la HPP del lactante se solapa frecuentemente con la forma perinatal letal. Las dificultades respiratorias derivadas de las alteraciones torácicas predisponen al desarrollo de neumonía e insuficiencia respiratoria y constituyen la principal causa de muerte1,2. La craneosinostosis puede conllevar aumento de la presión intracraneal, que puede asociarse a la aparición de la malformación de Arnold Chiari tipo i, hidrocefalia e hidrosiringomielia, entre otras anomalías neurológicas1,2,5,6. La hipertensión intracraneal y la calcificación de los ganglios basales pueden influir, junto con la alteración del metabolismo del PLP, en la fisiopatología de las convulsiones características de la HPP perinatal y del lactante, constituyendo un signo de mal pronóstico con elevada tasa de mortalidad5,11,13.
En la HPP del lactante es común observar niveles bajos de hormona paratiroidea. Su reducida secreción podría ser consecuencia de la hipercalcemia y determinar la aparición de hiperfosfatemia. Adicionalmente, la hipercalcemia e hipercalciuria pueden inducir el desarrollo de nefrocalcinosis, recomendándose incluir ecografías renales periódicas en el seguimiento14.
A pesar del mal pronóstico de esta forma de HPP, algunos casos han manifestado un incremento espontáneo en la mineralización ósea y una consiguiente mejoría clínica. Estos pacientes pueden presentar complicaciones neurológicas asociadas a la craneosinostosis, talla baja y pérdida prematura de la dentición, pero su pronóstico es más favorable que la forma neonatal5,11.
Las manifestaciones de la HPP infanto-juvenil se presentan como un continuum de las de la forma del lactante, apareciendo mayoritariamente tras el primer año de vida1,2. Radiológicamente, destacan las características proyecciones cartilaginosas de las placas en la metáfisis (lenguas de radiolucencia), diferenciales entre la HPP y distintas formas de raquitismo y displasias metafisarias12. La HPP infanto-juvenil puede cursar más raramente con hipercalcemia e hipercalciuria leves, siendo los niveles de parathormona y vitamina D habitualmente normales (o ligeramente bajos en los casos con insuficiencia renal)11.
En todas las formas pediátricas de HPP siempre debe considerarse el riesgo de craneosinostosis, recomendándose un seguimiento neurológico periódico, incluyendo oftalmoscopia, hasta la adolescencia. También conviene controlar a los pacientes con manifestaciones exclusivamente dentales debido a la potencial aparición ulterior de otras alteraciones. Se ha indicado que la gravedad de la enfermedad se correlaciona con el número de piezas dentales perdidas prematuramente6 y que es inversamente proporcional a la edad de la primera pérdida dental.
El primer escalón diagnóstico ante la sospecha clínica de HPP debería ser la determinación de la actividad total de la FA. Los valores de referencia en niños deben ajustarse en función de la edad, el sexo y el ensayo utilizado (tabla 5). Debe tenerse en cuenta que en formas leves de HPP pueden observarse niveles de actividad de FA tan solo ligeramente reducidos o incluso en el límite inferior de la normalidad1. Además, existen múltiples condiciones que pueden cursar con una actividad total baja de la FA sérica (tabla 4), por lo que también deben considerarse el aumento de ciertos sustratos de la FA1. En niños afectados de HPP, la magnitud del aumento de los niveles plasmáticos de PLP se correlaciona con la gravedad de la enfermedad, siendo este el marcador de mayor sensibilidad diagnóstica1. Según la experiencia internacional acumulada, su valor predictivo positivo y negativo para HPP en la infancia es muy alto, aunque la ausencia de elevación del PLP plasmático, o de los otros sustratos de la FA cuya determinación es menos accesible (PEA, PPi), no permite descartar el diagnóstico de HPP. La medición de la densidad mineral ósea mediante absorciometría dual de rayos X demuestra la reducción del contenido mineral óseo, aunque no es imprescindible para el diagnóstico.
Intervalos de referencia (U/l) de la actividad de la fosfatasa alcalina total
| Ensayo | Abbott Architect24,25 | Beckman Coulter26,27 | Ortho Vitros26,28 | Roche Cobas26,29 | Siemens Vista26 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sexo | Femenino | Masculino | Femenino | Masculino | Femenino | Masculino | Femenino | Masculino | Femenino | Masculino |
| 0-14 días | 90-273 | 77-237 | 91-256 | 83-248 | 81,5-248,7 | |||||
| 15 días-< 1 año | 134-518 | 116-450 | 131-476 | 122-469 | 121,7-472,6 | |||||
| 1-<10 años | 156-369 | 135-320 | 151-342 | 142-335 | 141,8-336,4 | |||||
| 10-<13 años | 141-460 | 122-400 | 137-424 | 129-417 | 128,1-419,6 | |||||
| 13-<15 años | 62-280 | 127-517 | 52-243 | 109-449 | 66-262 | 124-474 | 57-254 | 116-468 | 55,5-255,2 | 114,9-471,3 |
| 15-<17 años | 54-128 | 89-365 | 46-110 | 77-317 | 59-126 | 91-339 | 50-117 | 82-331 | 48,7-116,3 | 80,9-333,2 |
| 17-<19 años | 48-95 | 59-164 | 41-82 | 50-142 | 54-96 | 64-158 | 45-87 | 55-149 | 43,1-86,1 | 53,2-149,1 |
En un paciente con manifestaciones clínicas indicativos de HPP y niveles bajos de actividad total de FA sérica (en ausencia de otras causas), con/sin sustratos elevados, es necesario descartar otras patologías más frecuentes cuyo fenotipo puede solaparse con la HPP (tabla 6).
Diagnóstico diferencial de la hipofosfatasia en la edad pediátrica
| Diagnóstico diferencial de la hipofosfatasia | |
|---|---|
| HPP perinatal | Osteogénesis imperfecta (formas graves como la tipo ii) Enanismo congénito Displasia campomélica Condrodisplasias con alteraciones de la mineralización ósea Hiperparatiroidismo grave |
| HPP del lactante e infanto-juvenil | Alteraciones congénitas del metabolismo energético Aciduria orgánica Raquitismo primario y secundario Osteogénesis imperfecta Acondrodisplasia Periodontitis Displasia cleidocraneal Síndrome de Cole-Carpenter Osteoporosis idiopática juvenil Osteodistrofia renal Osteomielitis crónica no bacteriana Osteosarcoma Hipercalcemia idiopática y otras causas de nefrocalcinosis Negligencia y traumatismo no accidental |
Bien para establecer el diagnóstico definitivo, bien ante la ausencia de un diagnóstico diferencial claro, el hallazgo de mutaciones patogénicas mediante el estudio del gen ALPL permite establecer el diagnóstico de HPP15,16. Sin embargo, existen autores que postulan que, ante una sospecha clínica nítida (fenotipo+radiología+hallazgos analíticos), no es imprescindible la constatación de la alteración molecular para el diagnóstico de HPP1.
En mujeres embarazadas en las que se han detectado anomalías fetales compatibles con HPP, o con hijos afectados, puede realizarse estudio molecular en muestras de amniocentesis, aunque su utilidad es controvertida, ya que no puede diferenciarse de forma fiable entre HPP perinatal letal y prenatal benigna hasta la fase final de la gestación1,2.
Tratamiento de la hipofosfatasiaEl tratamiento debe ser multidisciplinar e individualizado según las características y gravedad de las manifestaciones clínicas. Los pacientes deberían seguirse en centros de referencia en el manejo de enfermedad del metabolismo óseo. Pese a que no existe un tratamiento curativo para la HPP, actualmente se dispone de terapia de reemplazo enzimático9,17.
Por su sintomatología y potencial mal pronóstico, la HPP perinatal grave presenta peculiaridades de tratamiento, por lo que a continuación se presenta un apartado específico (el resto de apartados hacen referencia al tratamiento de todas las formas pediátricas de HPP).
Hipofosfatasia perinatal graveEn primer lugar, en caso de sospecha prenatal se aconseja trasladar a la madre a un hospital con Unidad de Neonatología de nivel iii B-C.
En la HPP perinatal grave es muy importante el soporte respiratorio por la posibilidad de desarrollar hipoplasia pulmonar con insuficiencia respiratoria, que al nacimiento requiere generalmente intubación con ventilación mecánica. Puede precisarse ventilación de alta frecuencia, óxido nítrico, fármacos (sildenafilo, milrinona) e incluso oxigenación extracorpórea.
Deben controlarse estrechamente la FA, el calcio, el fósforo y la función renal cada 48-72 h en la primera semana de vida, y semanalmente después según evolución. Deberá también realizarse ecografía cerebral en las primeras 48 h y valorar evolutivamente según la clínica.
Tras la fase aguda inicial, si se requiere soporte respiratorio prolongado, se aconseja valorar si hay traqueobroncomalacia, que implicará soporte respiratorio con presión espiratoria elevada.
La función cerebral debe monitorizarse mediante electroencefalografía integrada por amplitud para detectar crisis convulsivas que pueden no tener correlato clínico y deben confirmarse mediante electroencefalografía convencional. Para su tratamiento, se administrará piridoxina i.v. (dosis inicial de 100mg; posteriormente, 15-30mg/kg/día en 3 dosis/día).
Tratamiento nutricional y de soporte (en cualquier edad)En caso de hipercalcemia, deberá restringirse la ingesta dietética de calcio y asegurar una correcta hidratación. En casos graves, puede considerarse el tratamiento con glucocorticoides o diuréticos, si bien el empleo de diuréticos del asa debe ser cuidadoso, puesto que pueden empeorar la hipercalciuria. Si los niveles plasmáticos de calcio y fosfato son normales, o mínimamente elevados, puede seguirse una dieta normocalcémica, ya que su suplementación puede emporar la hipercalcemia y la hipercalciuria. Asimismo, habría que evitar el empleo de suplementos de vitamina D3, excepto en situación carencial (sin necesidad de restringir la exposición solar), ya que la suplementación de calcio y/o vitamina D3 en estos casos se ha relacionado con hipercalcemia, hiperfosfatemia e hipercalciuria.
La salud dental debe vigilarse manteniendo controles periódicos y una correcta higiene oral.
El impacto funcional de la HPP está determinado principalmente por la afectaciones musculoesqueléticas, neurológicas y pulmonares, por lo que es importante la participación de médicos rehabilitadores y fisioterapeutas en el tratamiento de estos niños.
Tratamiento médicoNo existe un tratamiento médico único para los pacientes con HPP, habiéndose empleado diferentes estrategias con resultados variables.
Con el objetivo de disminuir la resorción ósea se ha empleado calcitonina, sin buenos resultados. Los bisfosfonatos pueden ser perjudiciales en la HPP18. Al ser análogos del PPi, contribuyen a inhibir la formación de hidroxiapatita y, por su capacidad de unión a cationes de cinc y magnesio, pueden inhibir la eventual actividad residual de TNSALP.
Los antiinflamatorios no esteroideos pueden ser de utilidad para el control del dolor, especialmente por edema óseo, y para el tratamiento de las calcificaciones extraarticulares18.
El reemplazo enzimático se ensayó inicialmente mediante la administración de plasma sanguíneo procedente de pacientes con enfermedad de Paget o purificados de placenta humana. Pese a cierta mejoría bioquímica, clínica y radiológica en algún paciente, esta fue transitoria y los resultados no fueron reproducibles. Para que este tratamiento sea efectivo no es suficiente con aumentar los niveles plasmáticos de TNSALP aisladamente, sino que esta debe poder incorporarse a la estructura ósea. Otras vías de sustitución que se han explorado son el trasplante de médula ósea o de células madre mesenquimales alogénicas determinadas hacia osteoblastos. Aunque se ha observado cierta mejoría clínica, debe tenerse en cuenta la necesidad de inmunosupresión y su morbilidad. También se ha intentado el tratamiento mediante transfección con terapia génica lentiviral en ratones TNSALP–/–18.
Desde 2015, la asfotasa alfa cuenta con aprobación de la Agencia Europea del Medicamento para el tratamiento de las formas de HPP perinatal, infantil y grave de la infancia19. Es una proteína de fusión formada por el dominio catalítico de TNSALP unido a una fracción constante de IgG1 con un decaaspartato terminal que aumenta su afinidad por el hueso. En 2012 se publicaron los primeros resultados terapéuticos con asfotasa alfa en HPP perinatal o infantil grave. Se observó una importante mejoría esquelética, de la función pulmonar, del desarrollo motor y cognitivo y de la miopatía, así como una rápida disminución de los niveles plasmáticos de PLP y PPi9, confirmándose dichos hallazgos tras un periodo más prolongado de tratamiento en un ensayo posterior17. Asfotasa alfa aumenta la supervivencia de los pacientes con HPP (incrementos del 27 al 84% a los 5 años10), especialmente en pacientes con necesidad de ventilación mecánica o con convulsiones vitamina B6 dependientes (aumento de supervivencia del 5 al 76% y del 0 al 77%, respectivamente)10.
Asfotasa alfa se administra por vía subcutánea en dosis habituales de 6mg/kg/semana en 3 o 6 dosis10,19. Los efectos adversos más frecuentes son reacciones locales en el punto de inyección, ocasionalmente con lipodistrofia local17. Se ha observado la aparición de anticuerpos específicos sin aparente repercusión en la efectividad clínica9. Es fundamental un correcto seguimiento, tanto para asegurar ajustes de dosis adecuados como para controlar posibles efectos adversos. Debe mantenerse la dosis ajustada al peso real del paciente, una dosis subóptima se relaciona con un rápido deterioro clínico y radiológico18. Debe vigilarse a la aparición de mineralización ectópica, manteniendo controles radiológicos, ecográficos renales y oftalmológicos. También puede ocurrir en territorio vascular18.
Tratamiento quirúrgicoDebe realizarse seguimiento de la posible afectación neurológica, especialmente la craneosinostosis. La indicación quirúrgica deberá considerarse en cada caso. El tratamiento con asfotasa alfa no parece ofrecer beneficio en este apartado9.
Asimismo, la cirugía puede ser necesaria para el tratamiento de fracturas o deformidades esqueléticas, como escoliosis. Una situación de descarga prolongada en caso de fractura podría ser factor de riesgo para fallo de consolidación18.
Conclusiones- –
La HPP es una enfermedad hereditaria rara del metabolismo óseo y mineral caracterizada por un déficit de actividad de la FA, que conlleva hipomineralización esquelética y dental y manifestaciones extraesqueléticas.
- –
El espectro fenotípico incluye desde formas precoces potencialmente mortales a formas más tardías, inespecíficas y menos sintomáticas.
- –
El diagnóstico en pacientes con alto índice de sospecha clínica debe incluir la constatación de niveles bajos de actividad sérica de FA, alteraciones radiológicas, la posible acumulación de sustratos de la enzima y el análisis molecular.
- –
La reciente aprobación de asfotasa alfa para el tratamiento de reemplazo enzimático en pacientes con HPP de inicio pediátrico supone un cambio en la estrategia terapéutica ante esta enfermedad.
Los autores declaran no tener ningún conflicto de intereses.
Los autores agradecen a Alexion Pharmaceuticals el apoyo logístico para la reunión del grupo de trabajo (los contenidos del cual son responsabilidad exclusiva de los autores) y el apoyo editorial por parte de Ogilvy Healthworld.
