


Caso clínico
Se solicitó consulta para una niña de 6,5 años por hipocrecimiento. Es hija de padres jóvenes, cuya talla genética (talla diana) es de 150,7 cm (1,8 desviación estándar [DE]) y tiene un hermano de 13 años que mide 144,6 cm (1,1 DE). La menarquia materna fue a los 15 años y no se refieren otros antecedentes familiares de interés, excepto que los abuelos, tres de ellos ya fallecidos (por accidente cerebrovascular, cáncer de mama y cáncer pancreático) eran de talla muy baja.
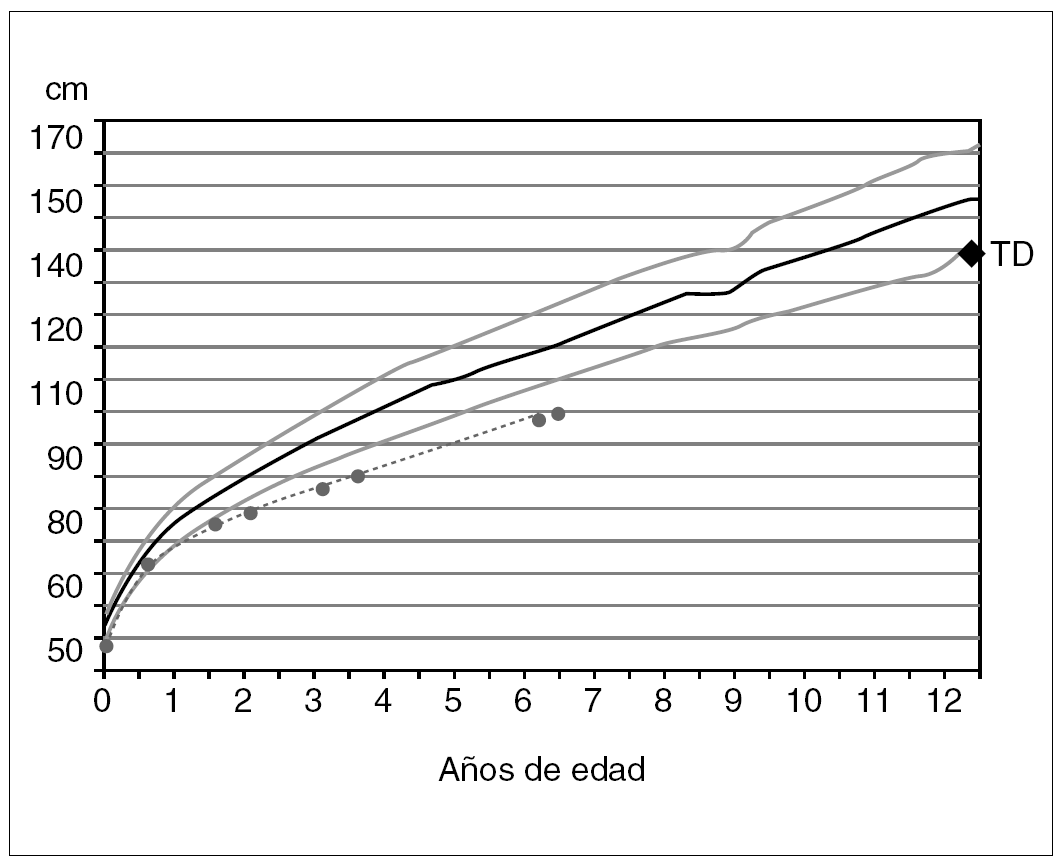
Durante su gestación, la madre fue tratada por diabetes gestacional. Nació a las 40 semanas, con 47 cm de longitud (1,9 DE) y 3.140 g de peso. Su crecimiento estatural (fig. 1) muestra una canalización inferior a la talla diana que se mantiene próxima a 3,3 DE desde los 3 años. Esta evolución corresponde con una velocidad de crecimiento entre 1,2 y 1,7 DE para su edad. A los 4 años, un accidente de tráfico le produjo un hundimiento frontoorbitario izquierdo que fue corregido quirúrgicamente. El desarrollo intelectual de la niña es normal. No ha sufrido trastornos nutricionales crónicos, ni se registra otro tipo de patología.
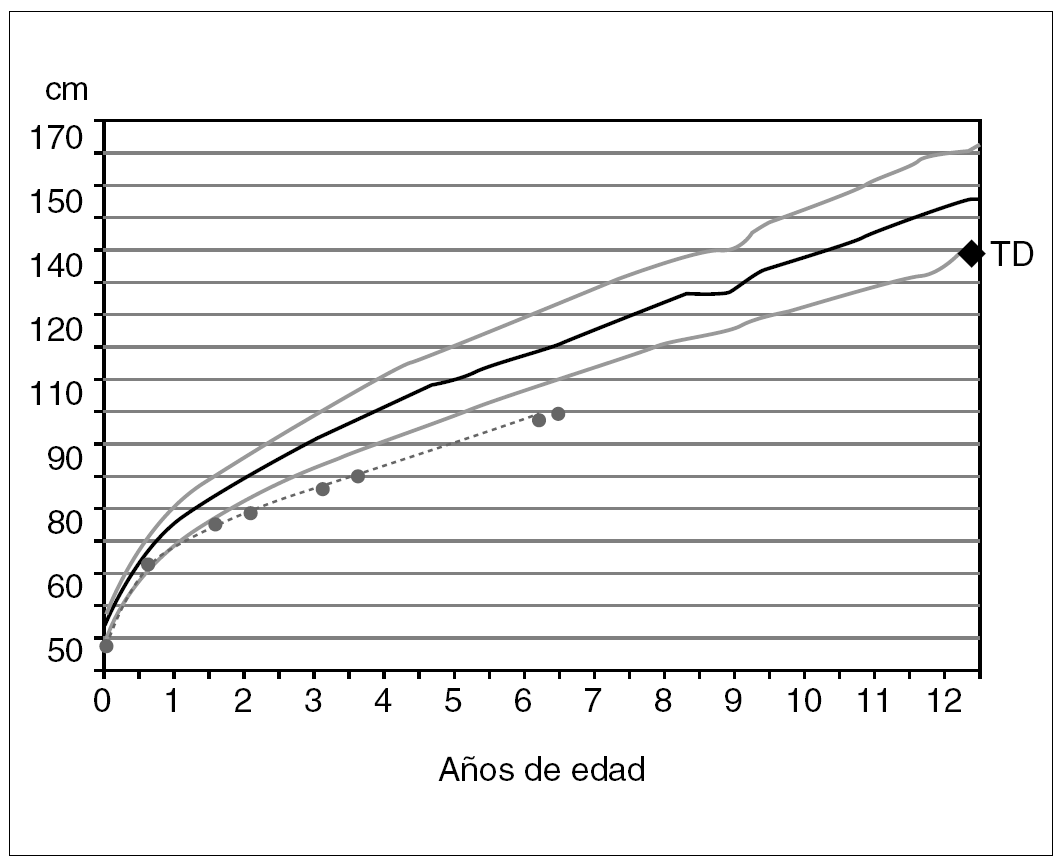
Figura 1. Evolución del crecimiento. TD: talla diana o genética (referencias auxológicas: Hernández et al. Fundación Orbegozo, 1988).
Su exploración muestra un fenotipo normal, sin displasias ni desproporciones aparentes, con una gran cicatriz frontotemporal izquierda, pero sin asimetrías faciocraneales. El resto de la exploración es normal, correspondiente a una niña impúber con psiquismo adecuado a su edad: talla, 100,6 cm (3,39 DE); peso, 17,5 kg (1,23 DE; 92,6 % para su talla); talla sentada, 54,8 cm (54,5 % de la talla); envergadura, 99,5 cm (98,9 % de la talla), relación rizo/mesomélica (longitud hombro-codo/codo-cabeza del tercer metacarpiano): 1,02.
El estudio complementario ofreció los siguientes datos:
Hemograma, bioquímica, análisis urinario, perfil celíaco y función tiroidea normales.
IGF-1: 108 ng/ml (1,8 DE), IGFBP-3: 2,47 μg/ml (0,03 DE). El estímulo con clonidina logró un pico de hormona de crecimiento en sangre de 17,2 ng/ml.
RM: imagen cerebral y área hipotálamo-hipofisaria normales.
Cariotipo en sangre periférica por bandas GTG: femenino normal, fórmula cromosómica 46,XX.
Edad ósea: 6,5 años (Greulich-Pyle). La radiografía practicada con este fin (fig. 2) muestra una imagen de características similares a la de su padre y su hermano (fig. 3).
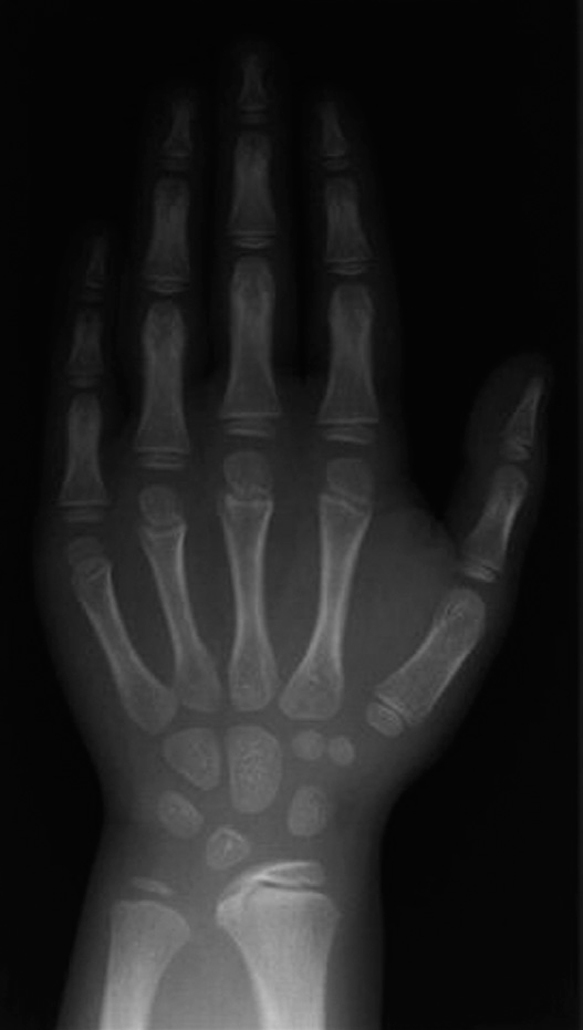
Figura 2. Radiografía de mano y muñeca solicitada para medir la edad ósea de la paciente.
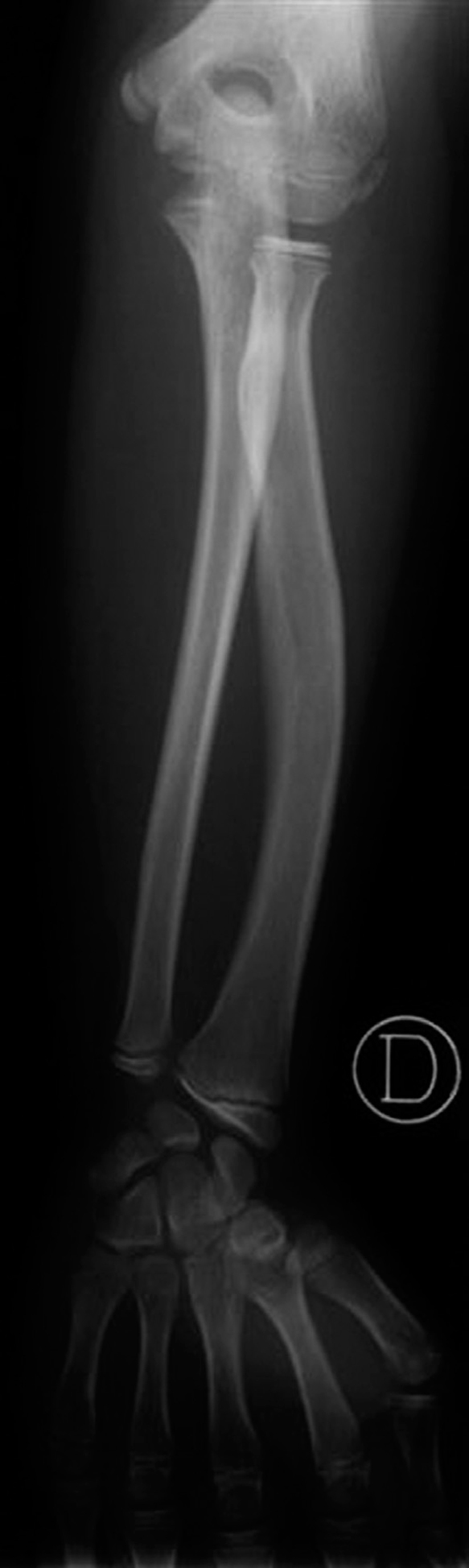
Figura 3. Radiología de antebrazo y muñeca del hermano, de 13 años.
Pregunta
¿Cuál es su diagnóstico?
Orientación: relacione los datos somatométricos con las imágenes radiológicas.
Discondrosteosis de Leri-Weill
La imagen de la radiografía de la paciente (fig. 2), muestra la displasia de Madelung de la Discondrosteosis de Leri Weill. La imagen, que solamente comprende carpo y mano, muestra varios signos, aún incompletos por la corta edad de la niña, que orientan al diagnóstico:
1.Deformación de la fila proximal del carpo, en la que el hueso semilunar se ha acercado a la epífisis radial, dando al borde carpal proximal, normalmente curvo, una configuración picuda (piramidal).
2.Distancia radiocubital ampliada (apreciable aquí, entre ambas epífisis).
3.Fusión precoz de la cara ulnar de la epífisis radial.
La figura 3, del antebrazo y muñeca del hermano púber, muestra la semiología displásica completa, sumando los siguientes signos a los ya referidos:
4.Incurvamiento con acortamiento radial.
5.Desviación ulnar de la superficie articular del radio.
6.Deformación triangular de la epífisis radial, cuyo borde externo es llamativamente más grueso que el interno.
A estos signos hay que añadir, en la mitad de los casos, la radiolucencia del borde ulnar distal del radio 1.
La deformidad de Madelung puede observarse también en niñas con síndrome de Turner (diagnóstico excluido aquí por el cariotipo), aunque sólo aparece en el 7 % de estas pacientes. El diagnóstico más sugerente para esta deformidad es la discondrosteosis de Leri-Weill (DLW), que la presenta en el 74 % de los casos y que en este caso se debe a deleción del gen SHOX (Short stature HOmeoboX-containing gene) en el 81 % de las ocasiones 2. Uno y otro diagnóstico pueden presentar, con similar frecuencia, otras anomalías esqueléticas, como cubitus valgus, paladar ojival y escoliosis.
La DLW también se asocia a talla baja, por acortamiento mesomélico de los miembros. No obstante este signo tiene expresión clínica muy variable, pues si la media de la talla de estos pacientes es de 2,2 DE, su rango alcanza de 4,6 DE a +0,6 DE. Esto significa que el 49 % de los pacientes con DLW tienen talla en rango normal (superior a 2,0 DE) 2.
La DLW, cuya incidencia absoluta varía entre 1/1.500 y 1/4.000 1 (comparable con el déficit de hormona del crecimiento o al síndrome de Turner), es más frecuente y grave en el sexo femenino, cuya estatura empeora 0,5 DE aproximadamente, con la estrogenización puberal 3. Todos los casos de DLW se deben a haploinsuficiencia (déficit monoalélico) del gen SHOX. Otros procesos tales como síndrome de Turner, displasia mesomélica de Langer (déficit homozigoto), una pequeña fracción (1-2 %) de niños con aparente talla baja idiopática, afectados de DLW 4 e incluso personas de talla normal, también son debidos a déficit de este gen. El gen SHOX codifica un factor de transcripción regulador del crecimiento del segmento medio de las extremidades (radio y tibia) y los dos primeros arcos faríngeos. Se localiza en la región seudoautosómica del brazo corto del cromosoma X (o su homóloga del Y) y, como sugiere este nombre, funciona como un gen autosómico. La DLW es la consecuencia de la deleción o anulación mutacional de uno de los alelos. La ubicación en esta región supone, además, la posibilidad de que la haploinsuficiencia SHOX forme parte del síndrome de genes contiguos del brazo corto distal del cromosoma X5. Es decir, un paciente con deleción del gen SHOX puede asociar a la DLW patologías tan dispares como condrodisplasia punctata, ictiosis ligada al X, síndrome de Kallmann, déficit mental, albinismo ocular 5 o incluso hemopatías malignas que pueden aparecer como complicación evolutiva de la DLW 6. Estas circunstancias y el evidente riesgo de engendrar un niño con la temible discondrosteosis homozigota (displasia de Langer) nos demuestran que el diagnóstico de DLW (incluyendo los que no tienen aspecto displásico y los de talla normal) trasciende al mero hipocrecimiento, por lo que se require el adecuado consejo genético.
A los pacientes con hipocrecimiento se les practica una radiografía de muñeca y mano, para medir su maduración ósea. La misma imagen es útil para investigar la posible existencia de la semiología radiológica aquí mostrada. Esto ya es posible en el escolar (aún sin deformidad visible en el antebrazo), y es un valioso instrumento diagnóstico a pesar de su sensibilidad limitada. La eventual identificación positiva debe seguirse del insalvable cariotipo y de la investigación molecular del gen SHOX en el paciente y sus familiares directos.
Correspondencia: Dra. A.C. Rodríguez Dehli.
Servicio de Pediatría. Hospital Universitario Central de Asturias.
Celestino Villamil, s/n. 33006 Oviedo. España.
Correo electrónico: crdehli@yahoo.es
Recibido en febrero de 2005.
Aceptado para publicación en septiembre de 2005.
