



Sr. Editor:
El síndrome de Cushing es raro en niños. En algunas series pediátricas1 no se encontraron diferencias en la distribución por sexos ni en la gravedad del hipercortisolismo, aunque en un estudio reciente se observa un predominio en varones en la edad prepuberal2. La coincidencia de un hipertirodismo tras la cura del hipercorticismo, como en nuestro caso, es extraordinariamente infrecuente y nos hace realizar una especulación fisiopatológica de la relación causal.
Presentamos el caso de un paciente varón de 12 años y 7 meses remitido por su pediatra por obesidad desde hace 3 años y escasa velocidad de crecimiento. En los antecedentes personales constaba una amigdalectomía con 3 años, y una intervención de colesteatoma a los 7 años. Entre los antecedentes familiares consta que su madre era obesa e hipertensa.
En la exploración clínica inicial se observaron un peso de 81,5kg, talla de 145cm (−0,75 SDS), índice de masa corporal (IMC) de 38,7 (6,88 SDS) y presión arterial de 115/80mmHg. Presentaba obesidad generalizada de predominio troncular, acumulación grasa cervical y dorsal alta, acantosis nigricans en axilas y lipomastia (fig. 1). Tenía testes en escroto prepuberales (3ml) y pubarquia III. No presentaba bocio.

Las pruebas complementarias en ese momento demostraron una hiperinsulinemia (18,5 μU/ml) con glucemia normal e índice HOMA de 4,02 (normal, inferior a 3,6). Los lípidos y las hormonas tiroideas fueron normales, así como las gonadotropinas y la testosterona (prepuberales). Se observó una discreta elevación del cortisol basal: 284ng/ml (110–225ng/ml).
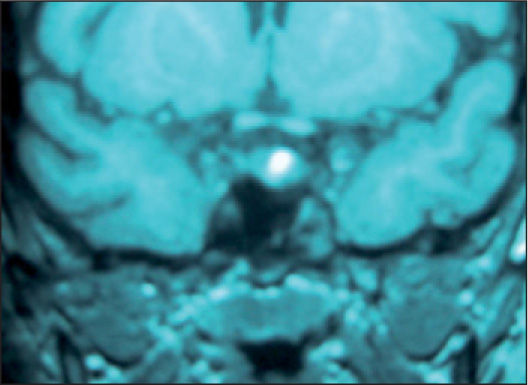
A los 6 meses había ganado peso (89,2kg) y la velocidad de crecimiento era nula. Los estudios del eje adrenal demostraron un cortisol libre urinario elevado (CLU): 131,8μg/24h (60μg/m2/24h), con discreta elevación de los valores de ACTH: 97pg/ml (20–72pg/ml) y cortisol normal (179ng/ml). Los andrógenos adrenales y mineralocorticoides fueron normales. En las pruebas de imagen la ecografía suprarrenal fue normal y en la resonancia magnética (RM) de hipófisis con contraste se observó un microadenoma hipofisario (fig. 2).
Ante estos resultados se efectuaron una serie de pruebas dinámicas: tests de supresión corto con 1mg de dexametasona: ACTH, 69pg/ml; cortisol, 164ng/ml; CLU, 81μg/24h; test de supresión largo con 2mg de dexametasona durante 3 días: ACTH, 97pg/ml; cortisol, 144ng/ml; CLU, 81μg/24h; con 8mg de dexametasona durante 3 días: ACTH, 54pg/ml; cortisol, 93ng/ml; CLU, 6,5μg/24h. Estos resultados confirmaron el diagnóstico de síndrome de Cushing dependiente de ACTH de origen hipofisario y se comenzó el tratamiento con ketoconazol, hasta la intervención quirúrgica transesfenoidal del adenoma. Los resultados de anatomía patológica fueron adenoma productor de ACTH.
Tras la intervención se mantuvo un tratamiento sustitutivo con hidrocortisona, y se normalizaron los valores hormonales: ACTH, 19pg/ml; cortisol, 35ng/ml; CLU, 12μg/24h.
A los 4 meses de la cirugía consultó por palpitaciones y nerviosismo. En la exploración física se observó una pérdida de peso (77,4kg) con talla de 148,2cm (6,46 standard deviation score [SDS] o puntuación z), taquicardia de 100 lat./min, sin bocio ni nódulos tiroideos. Pruebas complementarias: TSH: 0,00 μU/ml; T4L: 33,9 pmol/l; T3L: 13.52 pmol/l; anticuerpos antirreceptor de TSH y antiperoxidasa, negativos; ecografía tiroidea, tiroides de morfología y tamaño normal, sin nódulos. Se inició tratamiento con antitiroideos de síntesis durante 1 año con suspensión tras función tiroidea normal. Con anterioridad se había retirado la hidrocortisona tras la comprobación de una función suprarrenal normal.
En niños con escasa o nula velocidad de crecimiento, en especial en los que presentan obesidad o sobrepeso, es necesario descartar la existencia de un hipercortisolismo. El diagnóstico etiológico requiere determinaciones basales y pruebas dinámicas3. Entre las pruebas de imagen, la RM con contraste es el procedimiento más útil4. El tratamiento de elección es la cirugía transesfenoidal (CTE), con la cual se obtiene un porcentaje de curaciones del 70-90 % para los microadenomas y de menos del 60 % para los macroadenomas5.
El síndrome de Cushing endógeno, así como la administración exógena de glucocorticoides, pueden alterar el funcionamiento del eje hipotálamo-hipofisario-tiroideo. En adultos, la TRH se altera y la secreción pulsátil y nocturna de TSH se suprime en pacientes con enfermedad de Cushing6 y también tras la administración de glucocorticoides en voluntarios sanos y después de la infusión de hidrocortisona7; tras la resolución del hipercortisolismo puede aparecer o empeorar una enfermedad tiroidea autoinmune8.
En un trabajo reciente9 se encontró una mayor prevalencia de enfermedad tiroidea autoinmune, mayores tasas de hipotiroidismo subclínico y bocio en pacientes curados de un hipercortisolismo endógeno. Nuestro paciente desarrolló un hipertiroidismo primario con anticuerpos antirreceptor de TSH y anti-TPO negativos que no se positivizaron, por lo que es difícil establecer el origen de aquél, aunque creemos que puede tratarse de una forma de enfermedad autoinmune para la que no existe detección analítica, posiblemente por el método empleado10.
A pesar de la rareza de la enfermedad de Cushing en niños prepúberes, la aparición de una alteración tiroidea de probable origen autoinmune nos hace recomendar la realización de pruebas hormonales tiroideas tras el tratamiento de un hipercorticismo de cualquier origen.
