

La deficiencia de 11β-hidroxilasa (11β-OH) es la segunda causa más común de las formas clásicas de hiperplasia suprarrenal congénita (HSC) (5-8% de los casos). Clínicamente se caracteriza por virilización e hipertensión arterial. El objetivo de este estudio es describir las características clínicas, bioquímicas, genéticas y la evolución de los pacientes seguidos en nuestro centro.
Pacientes y métodosEstudio observacional longitudinal, retrospectivo y descriptivo. Criterios de inclusión: clínica de virilización, 11-desoxicortisol elevado en plasma y estudio del gen CYP11B1 con variantes patogénicas y probablemente patogénicas.
ResultadosSe identificaron 6 pacientes (1H, 5M) pertenecientes a 4 familias. La edad media al diagnóstico de los 4 pacientes índice fue de 2,3 años. Las pacientes 46,XX mostraron grados variables de virilización al diagnóstico, predominando el estadio 5 de Prader, con un caso de asignación de género masculino al nacimiento. Todos los pacientes presentaron aumento de las concentraciones séricas de 17-hidroxiprogesterona y testosterona. El 50% de los pacientes han desarrollado hipertensión arterial, con una mediana de edad de 9,3 años. Tres pacientes 46,XX han alcanzado talla final con una mediana de 154cm. Se encontraron 6 variantes diferentes distribuidas a lo largo del gen CYP11B1, 5 de las cuales son variantes no descritas previamente (c.595G>A, c.710T>C, c.1156delG, c.395+2dupT, c.1159dupA).
ConclusionesLos pacientes con formas clásicas de HSC por déficit de 11β-OH presentan una gran heterogeneidad en la expresión clínica. El diagnóstico y el tratamiento precoces son importantes para prevenir complicaciones y mejorar los resultados a largo plazo.
11β-hydroxylase (11β-OH) deficiency is the second most frequent cause of classic congenital adrenal hyperplasia (CAH) (5%-8% of cases). Clinically, it is characterized by virilization and arterial hypertension. The objective of this study was to describe the clinical, biochemical and genetic characteristics classic 11β-OH deficiency in patients managed in our hospital and its outcomes.
Patients and methodsRetrospective longitudinal, observational and descriptive study. Inclusion criteria: Patients with clinical features of virilization, high levels of 11-deoxycortisol and study of CYP11B1 gene with detection of pathogenic and likely pathogenic variants.
Resultswe identified 6 patients (1 male, 5 female) from 4 families. In the 4 index cases, the median age at diagnosis was 2.3 years. The 46,XX patients exhibited a variable degree of virilization at diagnosis, with a predominance of Prader stage V, and one case of male sex assignment at birth. All patients had elevated serum concentrations of 17-hydroxyprogesterone and testosterone. Fifty percent of the patients had developed arterial hypertension during the followup, with onset at a median age of 9.3 years. Three 46,XX patients reached a median final height of 154cm. Six different variants of the CYP11B1 gene were identified, 5 of which were novel variants (c.595G>A, c.710T>C, c.1156delG, c.395+2dupT, c.1159dupA).
ConclusionsThere is considerable heterogeneity in the clinical presentation of patients with CAH due to 11β-OH deficiency. Early diagnosis and treatment are important to prevent complications and improve long-term outcomes. We report 6 different variants of the CYP11B1 gene, including 5 novel variants.

La hiperplasia suprarrenal congénita (HSC) es un trastorno autosómico recesivo causado por defectos de distintas enzimas responsables de la esteroidogénesis suprarrenal, destacando la 21-hidroxilasa (21OHD), que representa el 90-95% de los casos1. El déficit de 11beta-hidroxilasa (11β-OH) es la segunda causa (5-8% de todos los casos), con una incidencia anual estimada de 1/100.000-200.000 nacidos vivos en poblaciones no consanguíneas, pudiendo llegar a 1/5.000 en la población judía marroquí2-5.
El déficit de 11β-OH es causado por alteraciones en el gen CYP11B1, localizado en el cromosoma 8q24.3. Comparte una alta homología con el gen CYP11B2 (aldosterona sintasa) separados por aproximadamente 40 kb6,7. Este gen codifica una enzima del sistema del citocromo P450, responsable de la conversión de 11-desoxicortisol a cortisol y 11-desoxicorticosterona (DOC) a corticosterona8,9. Hasta la fecha se han descrito alrededor de 180 variantes patogénicas, siendo la mayoría variantes con cambio de sentido (60%) y que tienden a agruparse en los exones 2, 6, 7 y 810-12.
El déficit de 11β-OH puede presentarse como fenotipo clásico o no clásico según el grado de gravedad clínica y el porcentaje de perdida de actividad enzimática. La forma clásica y más grave se debe a alteraciones severas con una actividad residual <10% que conduce a una disminución de la síntesis de cortisol con el consiguiente aumento de ACTH, sobreproducción de precursores de glucocorticoides y mineralocorticoides próximos al bloqueo y una biosíntesis excesiva de andrógenos3,13. Como resultado del hiperandrogenismo, las mujeres presentan grados variables de virilización de genitales externos, los hombres macrogenitosomia y todos pueden presentar pubertad precoz periférica, cierre epifisario prematuro y crecimiento somático acelerado. El acúmulo de DOC y sus metabolitos, que tienen acción mineralocorticoide, produce una retención de sodio y expansión de volumen que causa hipertensión hiporreninémica en aproximadamente 2tercios de los pacientes14,15.
A diferencia de los estudios clínicos de 21OHD, los estudios que informan sobre el fenotipo clínico y hormonal de la deficiencia de 11β-OH son limitados y se cuenta con pocas series publicadas debido a la rareza de esta enfermedad.
El objetivo de este estudio es describir las características clínicas, bioquímicas, genéticas y la evolución de pacientes con déficit de 11β-OH forma clásica en nuestro centro.
Pacientes y métodosEstudio retrospectivo observacional de una cohorte de pacientes con diagnóstico de déficit de 11β-OH forma clásica de la unidad de endocrinología pediátrica de hospital de tercer nivel durante periodo comprendido entre junio de 1996 y enero del 2022.
Criterios de inclusión: pacientes con hallazgos clínicos de virilización, niveles elevados de 11-desoxicortisol (> 7,2 ng/ml) y estudio del gen CYP11B1 con variantes patogénicas o probablemente patogénicas. Criterios de exclusión: no disponer de datos de seguimiento.
Registro de datos al diagnóstico y durante el seguimiento: edad cronológica (años), género asignado al diagnóstico, antecedentes familiares relevantes, consanguinidad, manifestaciones clínicas, hallazgos a la exploración física, resultados de laboratorio, radiología, dosis de hidrocortisona (mg/m2/día), medicación antihipertensiva y complicaciones asociadas. Los datos antropométricos se expresaron como desviación estándar (DE) utilizando datos de referencia españoles16. El grado de virilización genital de los pacientes 46XX fue estadificado según la clasificación de Prader17. La longitud del pene (LP) se expresó en cm y percentiles18. La pubertad se determinó según la estadificación de Marshall y Tanner19,20. La edad ósea se estudió mediante el método de Greulich y Pyle y la talla media parenteral (TMP) se calculó mediante el método de Tanner21. Se consideró diagnóstico de hipertensión arterial (HTA) cuando los valores repetidos de presión arterial en 3visitas separadas fueron mayores que el percentil 95 para la edad, sexo y talla del paciente22. Se realizó medida ambulatoria de la presión arterial (MAPA) mediante dispositivo CUSTO SCREEN PEDIATRIC en los menores de 12 años y CUSTO SCREEN 310 en los mayores de 12 años y analizado con el software CUSTO DIAGNOSTIC. Se realizó el análisis siguiendo las guías actuales de interpretación de MAPA en pediatría23.
El análisis estadístico se realizó como estadística descriptiva utilizando SPSS® versión 21.0. Se reportaron los valores como mediana y rango intercuartílico (IQR).
ResultadosSe identificaron 6 pacientes (1 H, 5M) procedentes de 4 familias diferentes: la familia 1 de etnia gitana y padres consanguíneos, la familia 2 de origen marroquí y padres no consanguíneos y los padres de los pacientes 3 y 4 de etnia caucásica no consanguíneos.
En ninguna de las familias se documentaron muertes neonatales ni antecedentes familiares endocrinológicos relevantes.
Características clínicas y bioquímicas al diagnósticoLas características clínicas al diagnóstico se muestran en la tabla 1. En los 4pacientes índice, la mediana de la edad de diagnóstico fue 2,3 (IQR 1,14-6,45) años. Los otros 2casos, hermanos menores de las familias 1 y 2, fueron diagnosticados a la edad de 7 meses (padres rechazaron diagnóstico prenatal) y en periodo prenatal, respectivamente. Ninguno de los 2recibió tratamiento prenatal con dexametasona por decisión parental.
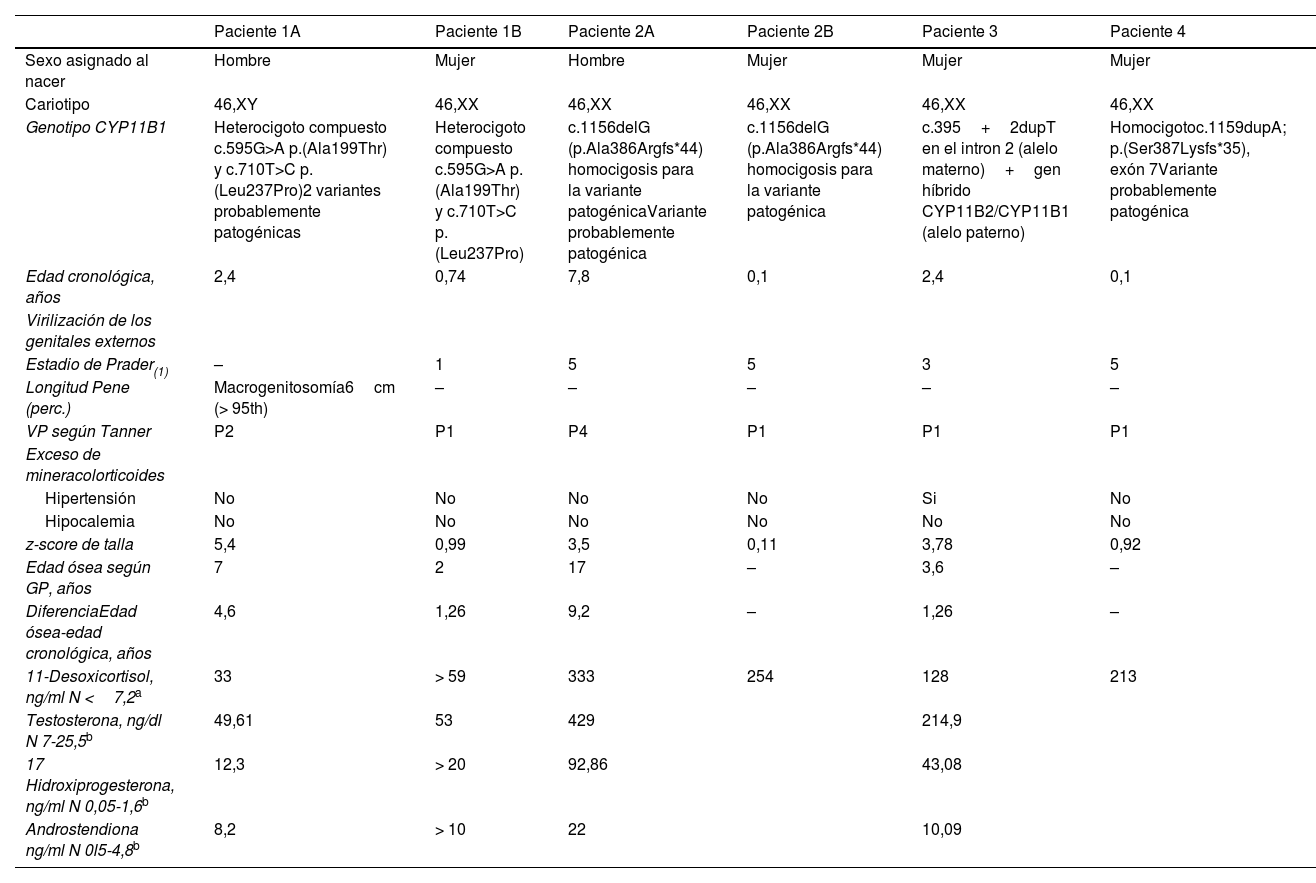
Características clínicas de los pacientes con déficit de 11β-OH en el momento del diagnóstico
| Paciente 1A | Paciente 1B | Paciente 2A | Paciente 2B | Paciente 3 | Paciente 4 | |
|---|---|---|---|---|---|---|
| Sexo asignado al nacer | Hombre | Mujer | Hombre | Mujer | Mujer | Mujer |
| Cariotipo | 46,XY | 46,XX | 46,XX | 46,XX | 46,XX | 46,XX |
| Genotipo CYP11B1 | Heterocigoto compuesto c.595G>A p.(Ala199Thr) y c.710T>C p.(Leu237Pro)2 variantes probablemente patogénicas | Heterocigoto compuesto c.595G>A p.(Ala199Thr) y c.710T>C p.(Leu237Pro) | c.1156delG (p.Ala386Argfs*44) homocigosis para la variante patogénicaVariante probablemente patogénica | c.1156delG (p.Ala386Argfs*44) homocigosis para la variante patogénica | c.395+2dupT en el intron 2 (alelo materno)+gen híbrido CYP11B2/CYP11B1 (alelo paterno) | Homocigotoc.1159dupA; p.(Ser387Lysfs*35), exón 7Variante probablemente patogénica |
| Edad cronológica, años | 2,4 | 0,74 | 7,8 | 0,1 | 2,4 | 0,1 |
| Virilización de los genitales externos | ||||||
| Estadio de Prader(1) | – | 1 | 5 | 5 | 3 | 5 |
| Longitud Pene (perc.) | Macrogenitosomía6cm (> 95th) | – | – | – | – | – |
| VP según Tanner | P2 | P1 | P4 | P1 | P1 | P1 |
| Exceso de mineracolorticoides | ||||||
| Hipertensión | No | No | No | No | Si | No |
| Hipocalemia | No | No | No | No | No | No |
| z-score de talla | 5,4 | 0,99 | 3,5 | 0,11 | 3,78 | 0,92 |
| Edad ósea según GP, años | 7 | 2 | 17 | – | 3,6 | – |
| DiferenciaEdad ósea-edad cronológica, años | 4,6 | 1,26 | 9,2 | – | 1,26 | – |
| 11-Desoxicortisol, ng/ml N <7,2a | 33 | > 59 | 333 | 254 | 128 | 213 |
| Testosterona, ng/dl N 7-25,5b | 49,61 | 53 | 429 | 214,9 | ||
| 17 Hidroxiprogesterona, ng/ml N 0,05-1,6b | 12,3 | > 20 | 92,86 | 43,08 | ||
| Androstendiona ng/ml N 0l5-4,8b | 8,2 | > 10 | 22 | 10,09 |
Prader 1954: 1: Hipertrofia de clítoris exclusivamente; 2: Hipertrofia de clítoris, fusión posterior de labios menores; 3: Hipertrofia de clítoris mayor, fusión casi completa de labios menores: Seno urogenital común; 4: Hipertrofia de clítoris importante, Seno urogenital común, labios mayores escrotalizados; 5: Genitales de apariencia masculina.
DE: desviación estándar; GP: Greulich and Pyle; N: normal; Perc: percentil; VP: vello púbico.
El paciente 46,XY se manifestó con macrogenitosomía y pubarquia precoz. Las 5 pacientes 46,XX mostraron grados variables de virilización al diagnóstico, 3 de ellas presentaron estadio 5 de Prader y un caso se asignó género masculino al nacimiento. Posteriormente, tras el diagnóstico de HSC a los 7,8 años de edad, los padres rechazaron la reasignación de género.
Al diagnóstico la edad ósea se encontraba avanzada, con una mediana de la diferencia edad ósea-edad cronológica de 2,9 (IQR 1,2-8,05) años. Todos los pacientes presentaron concentraciones séricas elevadas de 11-desoxicortisol, 17-hidroxiprogesterona (17OHP) y testosterona. Solo 1 paciente presento HTA al diagnóstico.
Hallazgos genéticosEn todos los pacientes índice se realizó estudio del gen CYP21A2 inicialmente, sin mostrar variantes patogénicas.
En el estudio del gen CYP11B1 (NM_000497.4) se encontraron en 3 familias variantes patogénicas no descritas en la literatura previamente.
En la familia 1 se observaron los cambios c.595G>A p.(Ala199Thr) (que afecta el último nucleótido codificante del exón 3, por lo que muy probablemente afecta al splicing; ESEfinder y otros programas (polyphen, SIFT) lo consideran patológico) y c.710T>C p.(Leu237Pro) que diferentes programas (polyphen, SIFT, mutationtaster) lo consideran patológico. En la familia 2 se documentó el cambio c.1156delG (p.Ala386Argfs*44) en homocigosis, cambio tampoco descrito y que, al tratarse de una deleción que da lugar a un cambio en el marco de lectura, puede predecirse que la proteína queda truncada y por lo tanto no funcional. En la paciente 3 se documentan 2 variantes en heterocigosis compuesta c.395+2dupT y gen híbrido CYP11B2/CYP11B1. La variante c.395+2dupT no ha sido descrita previamente en la literatura. En el paciente 4 se documentó un cambio en homocigosis en el exón 7 c.1159dupA; p.(Ser387Lysfs*35) que también da lugar a cambio en el marco de lectura.
Se realizó estudio genético a los padres en todos los casos y se documentó que eran portadores.
Tratamiento médicoTodos los pacientes recibieron tratamiento con hidrocortisona al diagnóstico de HSC con dosis media de 12,6 (IQR 12,02-14,4) mg/m2/día. Durante el curso del tratamiento, las dosis variaron entre 10 y 20mg/m2/día, salvo el paciente 2A requirió dosis más altas de hasta 27mg/m2/día para normalizar la presión arterial.
Tres pacientes (50%) recibieron tratamiento con fludrocortisona previo al diagnóstico de HSC por déficit de 11β-OH.
CirugíaEn 3 pacientes 46 XX, se realizó cirugía reconstructiva genital: 2 casos con estadio 5 de Prader y la otra, estadio 3. La mediana de edad de la primera cirugía correctora genital fue 10 (5-37) meses. Al paciente 2A se le realizó histerectomía, salpingooferectomía bilateral, resección de tercio superior de vagina y colocación de prótesis testicular a los 8 años de edad.
EvoluciónHan alcanzado talla final 3 pacientes.
El paciente 2A con cariotipo 46,XX género masculino y diagnóstico tardío a los 7,8 años alcanzó una talla adulta de 146cm (–2,81 DE con relación a la TMP).
Las otras 2pacientes que han alcanzado talla final fueron diagnosticadas en el periodo neonatal. La longitud al nacer en ambas pacientes fue normal para la edad gestacional, el z-score de la talla a los 3 años, al inicio de brote de crecimiento puberal (IBCP) y de la talla final fue de –1,2, –0,82 y –1,28 en la paciente 2B y de –1,04, –1,51 y –1,61 en la paciente 4. En función de la edad de IBCP se identificó que la paciente 2B fue maduradora temprana (edad de inicio 9 años) y la paciente 4 maduradora muy temprana (edad de inicio 8 años). Las 2pacientes presentaron un patrón de crecimiento con pérdida de talla en el primer año de vida, crecimiento prepuberal no recuperador y puberal normal. La talla final fue 1,02 y 0,55 DE por debajo de la TMP para la paciente 2B y 4, respectivamente.
Con relación al control de la presión arterial, 3 pacientes han presentado HTA, con una mediana de presentación de 9,3 (IQR 2,4-9,3) años. El paciente 2A presentó hipertensión a los 8 años del diagnóstico de HSC que se atribuyó a mala adherencia terapéutica, requiriendo tratamiento antihipertensivo con amlodipino. En los otros 2 pacientes (2B y 3) con HTA detectada en la consulta se ha realizado MAPA; al paciente 2B se le diagnosticó HTA ambulatoria de predominio nocturno y se implementaron medidas dietéticas y ajustes del tratamiento hormonal. En el paciente 3 se detectó HTA ambulatoria severa sin afectación de órganos diana, por lo que se aumentó la dosis de hidrocortisona objetivándose mejoría franca de los registros con esa medida. No se ha iniciado otro tratamiento y mantiene ausencia de afectación de órganos diana en el seguimiento.
Por último, la evaluación de la densidad mineral ósea (DMO) de 2 pacientes (2A y 4) mostró una puntuación z normal.
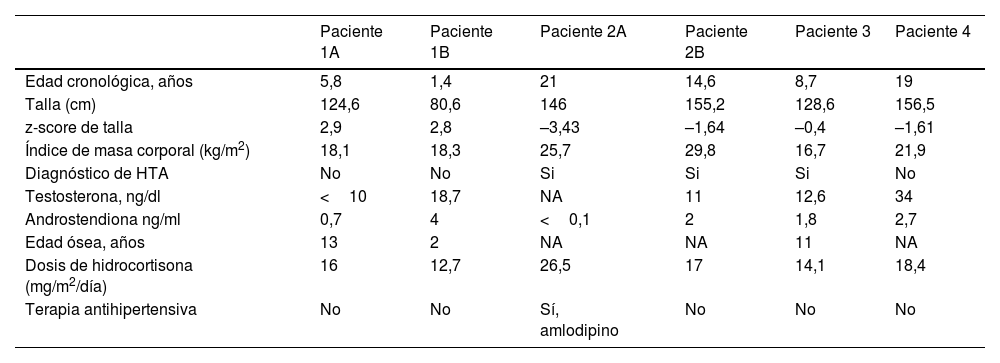
Las características clínicas en la última visita de seguimiento se muestran en la tabla 2.
Parámetros clínicos y bioquímicos de los pacientes con déficit de 11β-OH en la última visita de seguimiento
| Paciente 1A | Paciente 1B | Paciente 2A | Paciente 2B | Paciente 3 | Paciente 4 | |
|---|---|---|---|---|---|---|
| Edad cronológica, años | 5,8 | 1,4 | 21 | 14,6 | 8,7 | 19 |
| Talla (cm) | 124,6 | 80,6 | 146 | 155,2 | 128,6 | 156,5 |
| z-score de talla | 2,9 | 2,8 | –3,43 | –1,64 | –0,4 | –1,61 |
| Índice de masa corporal (kg/m2) | 18,1 | 18,3 | 25,7 | 29,8 | 16,7 | 21,9 |
| Diagnóstico de HTA | No | No | Si | Si | Si | No |
| Testosterona, ng/dl | <10 | 18,7 | NA | 11 | 12,6 | 34 |
| Androstendiona ng/ml | 0,7 | 4 | <0,1 | 2 | 1,8 | 2,7 |
| Edad ósea, años | 13 | 2 | NA | NA | 11 | NA |
| Dosis de hidrocortisona (mg/m2/día) | 16 | 12,7 | 26,5 | 17 | 14,1 | 18,4 |
| Terapia antihipertensiva | No | No | Sí, amlodipino | No | No | No |
NA: no aplica; HTA: hipertensión arterial,
En este estudio, describimos las características clínicas, bioquímicas, moleculares y la evolución a largo plazo de 6 pacientes con formas clásicas de HSC por déficit de 11β-OH.
En ausencia de programa de cribado neonatal, el diagnóstico de HSC por déficit de 11β-OH con frecuencia es muy tardío, especialmente en varones ya que no existe pérdida salina24. La edad al diagnóstico de HSC en nuestros 4pacientes índice fue de una mediana de 2,3 años, similar a la edad media de diagnóstico de 2,8 (rango 0-10) años reportada por Al-Jurayyan et al.25.
Con relación a la clínica, las 5 pacientes 46,XX mostraron grados variables de virilización al diagnóstico, 3 de ellas presentaron estadio 5 de Prader. En uno de nuestros pacientes se había asignado género masculino al nacimiento con un diagnóstico tardío de HSC. Algunos autores han descrito que en las mujeres con déficit de 11β-OH la virilización genital parece ser más severa que aquellas con déficit de 21-OH y que es más común que un recién nacido 46,XX con déficit de 11β-OH se le asigne el género masculino26,27. Destacamos que la paciente 1B de nuestro estudio no presentaba signos de virilización al nacimiento y posteriormente se presenta a los 8 meses con hipertrofia de clítoris.
El hiperandrogenismo adicionalmente se manifiesta en la etapa posnatal tanto en hombres como en mujeres como un rápido crecimiento somático y aceleración de la maduración ósea, que condiciona un cierre epifisario prematuro y talla baja adulta. Si bien hay una serie de estudios sobre la talla final en el déficit de 21-OH, hay muy pocos sobre el déficit de 11β-OH. Se ha observado una talla final alterada en el déficit de 11β-OH independientemente de la edad en el momento del diagnóstico, así como del control clínico y bioquímico3,15. Durante el seguimiento 3 de nuestros pacientes genéticamente 46,XX alcanzaron la talla final con una mediana de 154 (rango 146-154) cm y una mediana de la diferencia talla final-talla diana de -1,02 DE. Hochberg et al. reportaron que la talla adulta de 11 pacientes femeninas con déficit de 11β-OH de origen marroquí e iraní osciló entre los percentiles 3 y 5, con una media de 153,1±2,9cm, 10cm menos en comparación con la población general24. Por su parte Baş et al. refieren una talla final en los pacientes masculinos y femeninos de 1,5 DE inferior a la media10. El paciente 2A 46,XX con fenotipo masculino diagnosticado a una edad cronológica de 7,8 años alcanzó una talla adulta de 146cm (–2,81 DE de la TMP). En un estudio retrospectivo publicado recientemente, se evaluó a pacientes con HSC 46,XX con asignación de género masculina, 11 de los 38 pacientes que alcanzaron la talla final tenían déficit de 11β-OH y en ellos la mediana de la talla final fue de 149,2cm (rango 132,8-172)24.
En nuestra serie, 2de las pacientes que alcanzaron talla final fueron diagnosticadas en el periodo neonatal. Al analizar su crecimiento encontramos que, similar a lo que ocurre en la HSC por déficit de 21-OH, ambas pacientes presentaron una pérdida de talla en el primer año de vida posiblemente relacionado con las mayores dosis de hidrocortisona utilizadas28. Posteriormente presentaron un crecimiento prepuberal no recuperador, con un brote de crecimiento puberal normal (Z-score de talla al inicio del brote de crecimiento puberal igual al Z-score de talla final).
La HTA es una de las manifestaciones clínicas características del déficit de 11β-OH. Tradicionalmente se ha descrito hipertensión de leve a moderada en 2tercios de los casos en el momento del diagnóstico13,15; sin embargo, estudios recientes muestran que su frecuencia oscila entre el 53,6 y el 59%10,26. La hipertensión suele desarrollarse en la infancia y la adolescencia, aunque también puede desarrollarse durante los primeros años de vida11. En nuestra cohorte, 3 de nuestros pacientes desarrollaron HTA, 1 al diagnóstico y 2 a lo largo del seguimiento. El ajuste de la dosis de hidrocortisona permitió normalizar la presión arterial en 2 de ellos, mostrando cierta correlación entre el control metabólico y la HTA. Solo el paciente 2A ha requerido inicio de antihipertensivo que se ha atribuido a la pobre adherencia terapéutica y otros factores clínicos predisponentes como la obesidad. Se ha propuesto que la hipertensión se debe al exceso de DOC y sus metabolitos que tienen acción mineralocorticoide lo que lleva a retención de sodio y expansión de volumen y causa hipertensión hiporreninémica; sin embargo, no todos los pacientes desarrollan HTA y además no existe una correlación entre los niveles plasmáticos de DOC y la gravedad de la hipertensión26,29. Algunos autores proponen como causa de la HTA un posible aumento de la sensibilidad del tejido a DOC o la presencia de otros metabolitos con actividad mineralocorticoide más marcada25. Otro aspecto que se debe mencionar es que el grado de virilización y las características del exceso de mineralocorticoides tampoco están bien correlacionados. Algunas mujeres severamente virilizadas son normotensas, mientras que las pacientes levemente virilizadas pueden presentar hipertensión severa que conduce a complicaciones de órgano diana11,30. En nuestro estudio tampoco encontramos correlación entre el grado de virilización y la presencia de HTA. Al-Jurayyan et al. informaron de 4 hermanos con hallazgos de hipertensión e hipopotasemia con valores relativamente bajos de 11-desoxicortisol y leve virilización que persistieron a pesar de una adecuada terapia con hidrocortisona25.
Hasta la fecha, se han informado más de 180 variantes patogénicas en el gen CYP11B1 y la mayoría dan como resultado la forma clásica del déficit de 11β-OH10,30. A diferencia de la deficiencia de 21-hidroxilasa la correlación entre fenotipo y genotipo en el déficit de 11β-OH aún no está completamente establecida, ya que los estudios genéticos moleculares del déficit de 11β-OH son relativamente escasos y a menudo las variantes patológicas identificadas del gen CYP11B1 no se han caracterizado funcionalmente. Varios informes describen que la gravedad clínica y los hallazgos bioquímicos pueden diferir significativamente incluso dentro de miembros de la misma familia con el mismo genotipo, lo que indica una variabilidad fenotípica25,31. En nuestro grupo de estudio encontramos 6 variantes diferentes distribuidas a lo largo del gen CYP11B1, 5de las cuales son variantes no descritas previamente (c.595G>A, c.710T>C, c.1156delG, c.395+2dupT, c.1159dupA). En la paciente 3 se documentan 2 variantes en heterocigosis compuesta c.395+2dupT y gen híbrido CYP11B2/CYP11B1. La variante c.395+2dupT no ha sido descrita previamente en la literatura. Según el programa «in silico» ESE-Finder este nuevo cambio afecta a la secuencia dadora del splicing. Se han descrito otros cambios parecidos causantes de la enfermedad; en 2018 Baş et al. describieron la variante c.395+3A>G en un paciente masculino10. Hasta la fecha son pocos los pacientes en los que se ha informado que presentan el gen quimérico CYP11B2/CYP11B132-35. Esto fue descrito por primera vez en 2001 por Portrat et al., los cuales identificaron un gen quimérico homocigoto que contenía el promotor y los exones 1-6 de CYP11B2 y los exones 7-9 de CYP11B132. Serían necesarios realizar estudios funcionales para demostrar los mecanismos patogénicos de dichos cambios.
En conclusión, el déficit de 11β-OH es una causa rara de HSC. Los pacientes con forma clásica presentan una gran heterogeneidad en la expresión clínica sin una clara relación entre genotipo y fenotipo, a diferencia del déficit de 21-OH. El diagnóstico y el tratamiento precoces son importantes para prevenir complicaciones y mejorar los resultados a largo plazo. Además, informamos 6 variantes diferentes en el gen CYP11B1, incluidas 5 variantes no descritas previamente y su correlación con los hallazgos clínicos y bioquímicos, ampliando el espectro de variantes patogénicas del gen CYP11B1 y contribuyendo así a mejorar el asesoramiento genético.
Declaración sobre el uso de la IA generativa y de las tecnologías asistidas por la IA en el proceso de redacciónLos autores declaran que no se ha usado IA generativa y tecnologías asistidas por la IA en el proceso de redacción.
FinanciaciónLa presente investigación no ha recibido ayudas específicas provenientes de agencias del sector público, sector comercial o entidades sin ánimo de lucro.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Este trabajo fue presentado como comunicación oral en el 44.° Congreso de la Sociedad Española de Endocrinología Pediátrica, celebrado en Oviedo del 11 al 13 de mayo del 2022.

