




El tumor de Wilms es el cuarto tumor más frecuente en pediatría, tras leucemias, tumores del sistema nervioso central y neuroblastoma. La edad típica de presentación es entre los 3 y los 4 años1. El signo más frecuente es el hallazgo incidental de una masa abdominal, siendo la hematuria macroscópica poco común como clínica de presentación. Existe una variante de nefroblastoma denominada botrioide, en la que este signo sí es frecuente debido a su crecimiento hacia el sistema colector renal2.
Presentamos el caso de un varón de 2 años, asintomático y sin antecedentes personales ni familiares de interés, que acude a urgencias por el hallazgo de coágulo en el pañal. La exploración física no aportó información relevante.
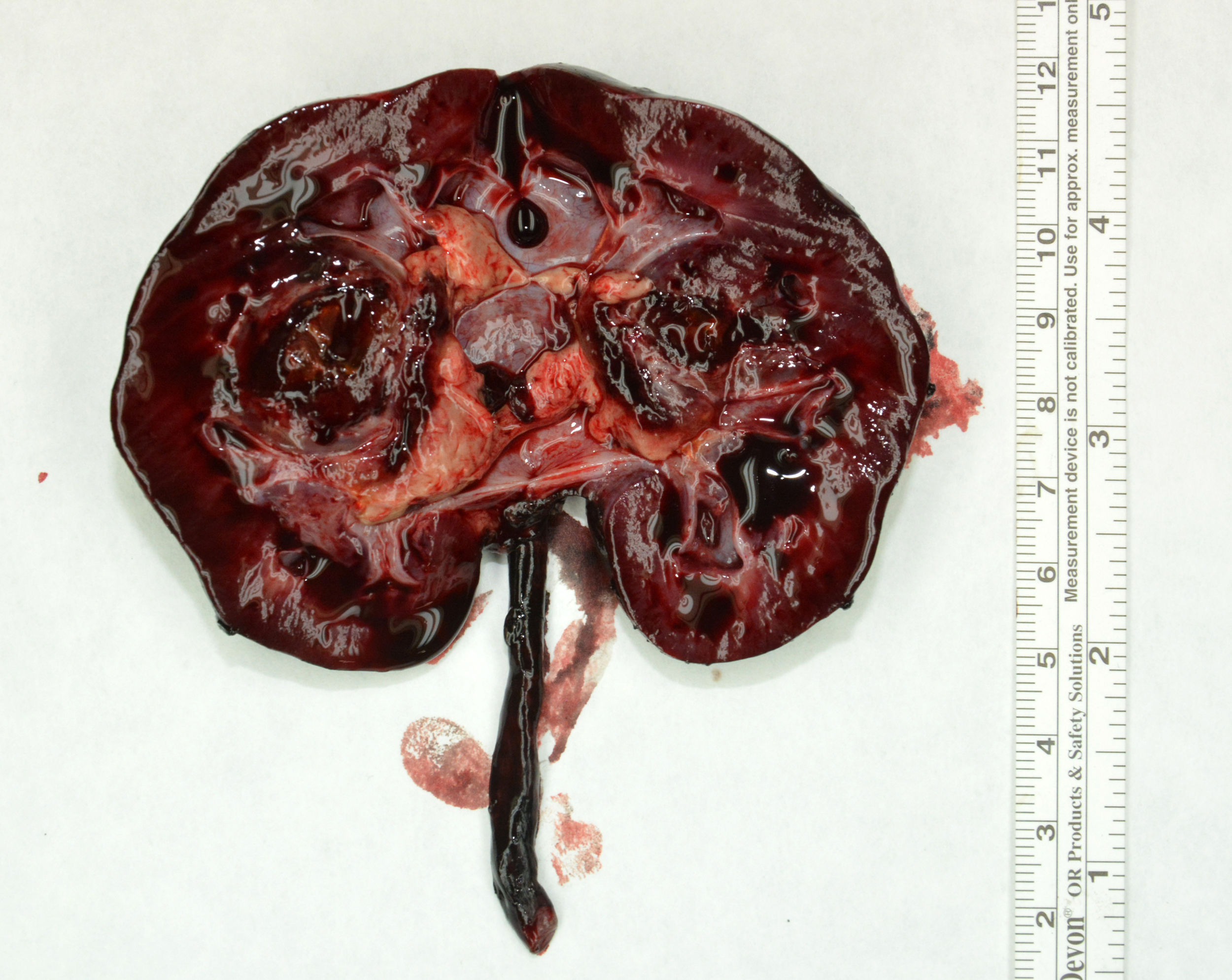
Se realizó ecografía abdominal donde se apreció una lesión focal en mesoriñón derecho con extensión a pelvis renal, asociando dilación calicial retrógrada. Se completó el estudio con una resonancia magnética con contraste, identificando una masa sólida en la pelvis renal derecha de 50×47×36mm en ejes transverso, craneocaudal y anteroposterior, que alcanzaba el tercio superior del uréter e infiltraba el parénquima del mesoriñón y del polo inferior renal. Heterogénea y levemente hiperintensa en las secuencias potenciadas en T2 e hiperintensa en T1. Restringía a la difusión y realzaba tras la administración de contraste. Los cálices renales derechos y el uréter ipsilateral estaban dilatados.
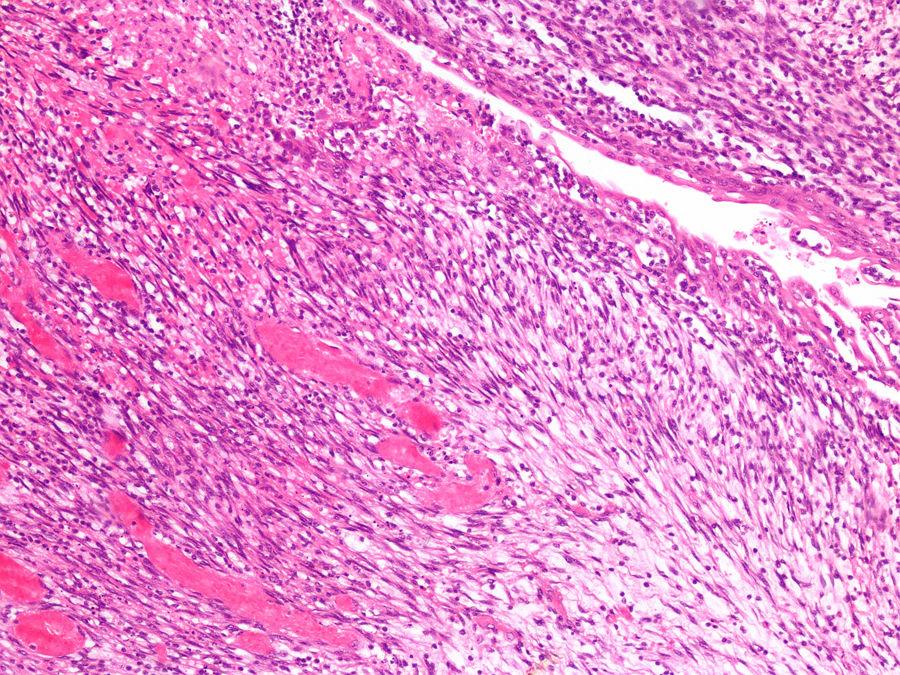
Debido a la dificultad de diagnóstico radiológico, se decidió realizar una biopsia percutánea ecodirigida con aguja de 16G. La anatomía patológica fue compatible con el tumor de Wilms. El estudio de extensión fue negativo. Recibió quimioterapia con 4 semanas de vincristina y actinomicina, y posteriormente se realizó nefrectomía derecha. El estudio histológico reveló nefroblastoma de tipo estromal estadio II por infiltración del seno renal (Imagen 1 y 2). Posteriormente se completó tratamiento con 28 semanas de vincristina y actinomicina. Actualmente se encuentra libre de enfermedad a los 18 meses de finalizar el tratamiento.
El 90% de los tumores renales en la infancia son nefroblastomas. Estos suelen localizarse en el parénquima renal. Los restos nefrogénicos (focos de células embrionarias que persisten más allá de la semana 36 de gestación), son considerados precursores del nefroblastoma ya que se han encontrado hasta en el 40% de los pacientes con este tumor, sin asociarse a otros tumores renales. Según su localización en el parénquima, se clasifican en perilobares e intralobares. Estos últimos implican un trastorno en una etapa más precoz del desarrollo, son menos frecuentes, pero presentan mayor asociación con la degeneración en tumor de Wilms botrioide3.
La realización de biopsia confirmatoria en los casos del tumor de Wilms con características clínicas y radiológicas típicas, no forma parte de la práctica habitual de los protocolos del Grupo de Estudio de Tumores Renales de la Sociedad Internacional de Oncología Pediátrica (SIOP-RTSG). En torno al 10% de los casos se presentan como una masa polipoidea, con aspecto de racimo de uvas, que invade el sistema colector, adquiriendo un patrón de crecimiento «botrioide». Esto suele conllevar la realización de biopsia para su confirmación histológica. En el caso de planificar una biopsia, la recomendación es hacerla percutánea, con aguja de 16 o 18G y evitando biopsias excisionales que puedan sobreestadificar el tumor.
En 1981 se introdujo el concepto de «tumor de Wilms botrioide» para referirse a este patrón de crecimiento4. Inicialmente se pensó que tenían peor pronóstico al considerar que se producía una invasión del sistema colector. Posteriormente, se demostró que en la mayoría de los casos no existe infiltración de la pared del sistema excretor5. Numerosos autores han abogado por considerarlo una variante de mejor pronóstico, debido al mayor porcentaje de histología favorable2, y a un diagnóstico precoz, al ser frecuente la hematuria macroscópica de forma temprana6. Sin embargo, los 2 principales grupos de estudio de tumores renales (Children Oncology Group e International Society of Paediatric Oncology) continúan sin reconocerlo como subtipo.
En cuanto a la resección del tumor de Wilms, la vía de abordaje abdominal es de elección. Para evitar la siembra tumoral ureteral o vesical, y más en tumores con localización pélvica, se recomienda la ligadura del uréter lo más distal posible a la pelvis renal y siempre de forma precoz, antes de cualquier manipulación tumoral.
Nos parece importante considerar el diagnóstico de tumor de Wilms ante cualquier paciente con una masa renal, incluso cuando la afectación sea exclusiva o mayoritaria del sistema colector. Actualmente el tratamiento y el pronóstico de los tumores de Wilms debe establecerse de acuerdo al subtipo histológico y al estadio, sin tener en cuenta el patrón de crecimiento botrioide2.
