
Introducción
La enfermedad de Ménétrier (EM), descrita por primera vez en 18881, es una rara entidad clínica de etiología desconocida, caracterizada, macroscópicamente, por la presencia de pliegues gástricos hipertróficos e, histológicamente, por el hallazgo de hiperplasia foveolar masiva con escasa inflamación y un marcado incremento del espesor de la mucosa. Suele asociarse a hipoproteinemia secundaria a la pérdida proteica hacia la luz gástrica.
La presencia de pliegues gástricos hipertróficos no presupone el diagnóstico de EM2, dado que existen otras entidades clínicas que también cursan con hipertrofia de los pliegues gástricos (tabla1). Estos procesos deberían englobarse bajo la denominación de "Gastropatía Hipertrófica" (incluyendo la EM).

El término "enfermedad de Ménétrier" debe reservarse para aquellos casos raros que cumplen totalmente la descripción original: "hiperplasia foveolar masiva sin gastritis".
El aumento de pérdidas proteicas gástricas puede hallarse ocasionalmente en muchas de estas enfermedades que están asociadas con pliegues gástricos engrosados, pero la pérdida proteica excesiva no es una característica universal de todos estos procesos; p.ej., no la presenta el síndrome de Zollinger-Ellison y su presencia en la enfermedad de Ménétrier es variable.
La EM en adultos suele presentar una sintomatología crónica y grave, la pérdida proteica no siempre está presente y, sólo el 25% de los pacientes tienen edemas. El inicio de los síntomas es insidioso, y las remisiones espontáneas son excepcionales. El curso habitual es prolongado, sin remisión y cerca de un 10% presentarán degeneración maligna.
La presencia de enteropatía pierde-proteínas junto con hipertrofia de pliegues gástricos en la infancia ha sido etiquetada como EM infantil. En los niños suele ser una situación benigna, transitoria y autolimitada, y constituye un proceso diferente de la EM del adulto (son diferentes el curso clínico, el pronóstico y probablemente la etiología). Aesta situación clínica se le conoce en la actualidad como Gastropatía hipertrófica pierde-proteínas en la infancia (GHPP), con unos 50 casos publicados hasta la actualidad.
Aspectos clínicos de la GHPP infantil3
La enfermedad suele presentarse en los primeros años de vida, con un predominio de varones. La aparición de edemas (a veces con ascitis y/o derrame pleural) constituye el hallazgo clínico fundamental. Muchos casos presentan síntomas prodrómicos inespecíficos (vómitos, diarrea, dolor abdominal, catarro de vías altas, malestar, fiebre, etc.) en ocasiones autolimitados y que han desaparecido al evidenciarse los edemas. En el 10% de casos se asocia una hemorragia gastrointestinal franca.
Todos los pacientes presentan hipoproteinemia e hipoalbuminemia y en más de la mitad de los casos se constata la presencia de eosinofilia periférica. Sólo se ha detectado anemia en los casos que han presentado hemorragia gastrointestinal evidente. En todos se descarta la existencia de enfermedad hepática y renal.
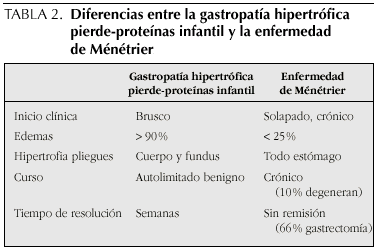
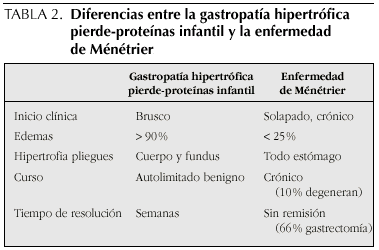
La gastropatía hipertrófica pierde-proteínas de la infancia tiene características similares a la EM del adulto, pero existen diferencias importantes3,4 que se reflejan en la tabla 2.

Las pérdidas proteicas por el tracto gastrointestinal pueden comprobarse mediante la medición de la a1-antitripsina en heces o por el test de Gordon; en ambos casos no se demuestra que la pérdida proteica tenga lugar en el estómago. Para ello puede recurrirse a la determinación del contenido proteico en jugo gástrico (proteínas totales o a1-antitripsina) o por gammagrafía abdominal tras inyección por vía endovenosa de albúmina-99 mTc y comprobar su acumulación en el tracto gastrointestinal, apareciendo primero en el estómago5.
El estudio radiológico baritado del tracto digestivo evidencia la existencia de pliegues gástricos agrandados, en fundus y cuerpo gástrico, a veces con aspecto irregular, polipoideo. El antro suele estar respetado y no hay afectación del intestino delgado. Al no existir patrones de normalidad del tamaño de los pliegues gástricos en pediatría, la interpretación de los pliegues engrosados suele ser bastante subjetiva. Además, en ocasiones no se comprueban endoscópicamente los hallazgos radiológicos6, por lo que debe darse un valor relativo a los mismos.
La ecografía abdominal puede demostrar la presencia de los pliegues gástricos hipertróficos7: se observa un engrosamiento de los pliegues de la mucosa con varias estructuras hiperecoicas en la capa situada debajo de la mucosa. Están engrosadas las capas hipoecoica (mucosa y submucosa) y la capa hiperecoica (eco interfase entre submucosa y muscular). En el fundus los pliegues engrosados adoptan el aspecto de "circunvalaciones cerebrales". Las características de la exploración (no invasiva e inocua) hacen que constituya un buen método para poder controlar la evolución local de la enfermedad.
La endoscopia comprueba la existencia en cuerpo gástrico y en fundus, de pliegues hipertróficos, edematosos-hiperémicos, recubiertos de un exudado de aspecto gelatinoso y a veces con erosiones superficiales de bordes irregulares, dendríticos. Los pliegues son duros al tacto y no desaparecen con la insuflación. Esófago, antro y duodeno tienen un aspecto macroscópicamente normal.
La histología se caracteriza por la presencia de una hiperplasia foveolar, junto con glándulas gástricas dilatadas, hipertróficas, rellenas de moco, edema e infiltrado inflamatorio (neutrófilos, eosinófilos y linfocitos y en ocasiones células plasmáticas). Para efectuar un diagnóstico histológico de certeza es necesario la biopsia de todo el grosor de la mucosa8. Las biopsias obtenidas con las pinzas convencionales del endoscopio suelen ser inadecuadas, ya que los hallazgos característicos se encuentran en la parte más profunda de la mucosa. También se han descrito casos con histología normal9, por lo que el diagnóstico se basa muchas veces en la combinación de datos: hipoproteinemia junto con la comprobación endoscópica de la hipertrofia de pliegues y las biopsias superficiales.
Las medidas terapéuticas adoptadas suelen ser de sostén (seroalbúmina, diuréticos, restricción de sal, dieta hiperproteica, nutrición parenteral, anti-H2 o inhibidores de la bomba de protones). Se describen 4 casos que precisaron resección gástrica para controlar la hemorragia.
La duración media de la enfermedad (inicio síntomas hasta desaparición de los edemas) suele ser de 5 semanas (rango, 2-18) aunque existe la excepción de un caso publicado por Faure10 con una evolución de más de 26 semanas.
Consideraciones etiológicas
La etiología de las gastropatías hipertróficas es desconocida; las diferencias vistas entre los casos pediátricos y los de adultos apuntan la posibilidad de que pueda ser multifactorial, o quizás representa una lesión final común producida por múltiples causas. En algunos casos8 parece existir una predisposición hereditaria autosómica dominante.
Se sabe que diversos estímulos pueden conducir a la hiperplasia gástrica focal; entre estos estímulos se han considerado toxinas, factores alimentarios, anomalías inmunológicas, afecciones autoinmunes, alergia y agentes infecciosos.
Las enfermedades infecciosas se habían ligado a la hipertrofia de pliegues gástricos desde hace tiempo, asociadas a sífilis e histoplasmosis; desde 1971se han publicado casos de GH pierde-proteínas infantil asociada a infección por citomegalovirus (CMV) y más recientemente asociados a infecciones por Helicobacter pylori (Hp).
En 1971, Lachman publicó 3 casos de GH pierde-proteínas en niños, uno de los cuales presentaba cuerpos de inclusión citomegálicos en las células glandulares gástricas; desde entonces se han ido publicando casos de GH pierde-proteínas infantil asociados a infección por CMV (alrededor de un 30% de los 50 casos publicados hasta la actualidad). Recientemente se ha publicado el primer caso en un adulto con GH pierde-proteínas asociado a infección por CMV, con la misma evolución que los casos infantiles11, por lo que los autores consideran que debe buscarse el CMV en los pacientes que presentan un inicio rápido de síntomas y pérdidas proteicas importantes por el tracto gastrointestinal.
La infección por el CMV puede ser: a)primaria; b)una reactivación de una infección latente, o c)una reinfección con una cepa antigénica diferente.
El diagnóstico de infección por CMV se comprueba mediante la identificación y aislamiento del virus en la mucosa gástrica: demostración de los cuerpos de inclusión intranucleares, cultivo, detección de antígenos virales, determinación del genoma del CMV por hibridación in situ o por técnicas de PCR.
Para diferenciar la primoinfección de las reactivaciones se suele emplear la serología, aunque los resultados sean difíciles de interpretar sobre todo en pacientes que inicialmente pueden presentar IgG bajas debido a las pérdidas proteicas; incluso las IgM-CMV, que en principio indicarían infección primaria, pueden también reaparecer durante la reactivación del CMV y no distinguir por lo tanto, entre infección primaria o reactivación de una infección latente.
La demostración de la presencia del virus la mucosa gástrica no descarta la posibilidad de que el CMV sea un invasor secundario en una mucosa lesionada y no el factor etiológico primario.
Una relación causal entre infección gástrica por CMV y GH pierde-proteínas se establece mejor detectando el virus en la mucosa gástrica y además comprobando la seroconversión y detección de IgM-CMV3.
En 1988, Lepore et al12 publican el primer caso de una paciente afectada de EM, en la que tras 23 años de evolución demuestran la presencia de Helicobacter pylori (HP). El tratamiento erradicador condujo a la curación de la paciente.
Desde entonces han aparecido un total de 6publicaciones13-18 con otros tantos pacientes adultos afectos de EM con evoluciones clínicas de 4 meses a 3 años, en los que se demostró la presencia de HP. El tratamiento erradicador (con diferentes pautas terapéuticas) produjo la curación clínica y la normalización de la mucosa gástrica en todos ellos, aunque en un caso la mejoría no apareció hasta transcurridos 12 meses después de finalizar dicho tratamiento17. Los diferentes autores llegan a la conclusión de que, al menos en un subgrupo de pacientes afectos de gastritis hipertrófica pierde proteína, HP es un factor patogénico y aquellos que se hallan afectados de una EM deben ser estudiados para descartar la infección por HP y deben ser tratados para erradicarlo en caso de comprobar su existencia.
No obstante, Simon et al19 publican el caso de un adulto con EM en el que no consiguen demostrar la presencia del HP, pero que el tratamiento con omeprazol, amoxicilina y claritromicina produce también la curación del paciente. Anteriormente, Raderer20 había publicado un paciente de características similares, sin infección por HP, en el que se mantiene una curación clínica, no histológica, durante 3 años con la administración ininterrumpida de tratamiento triple con lansoprazol, claritromicina y metronidazol. La mejoría en este caso estuvo ligada a la asociación claritromicina+metronidazol, ya que al cambiarlos por amoxicilina el paciente empeoró a pesar de mantener el lansoprazol.
Estas dos publicaciones apuntan la posibilidad de que la infección por HP sea una coincidencia en la EM. Además, el efecto beneficioso de los inhibidores de la bomba de protones no está estrictamente ligado al tratamiento erradicador21.
Se han publicado 6 niños en 4 trabajos22-25, asociando la presencia de infección por Hp con la GH pierde-proteínas infantil. En cuatro22,24,25 la curación se inicia, según los autores, tras el comienzo del tratamiento erradicador (eritromicina en tres y triple terapia con amoxicilina, metronidazol y lansoprazol en el cuarto), pero en un momento de la evolución clínica en que esta mejoría también podría ser atribuida al curso natural autolimitado del proceso; además, en uno de los casos no se consigue la erradicación del germen22.
En los 2 pacientes no tratados, la curación espontánea se produce a los 2 meses de evolución; en uno se comprueba el aclaramiento natural del germen22, mientras que en el otro, no sólo persistía tras la curación sino que habían empeorado las lesiones de gastritis crónica asociadas al HP.
Hasta el momento actual parece que la infección por HP como agente causal de la GH pierde-proteínas infantil sea muy poco probable y que el germen posiblemente es un mero "espectador" en la aparición y en la evolución del proceso en los niños.
En síntesis, la GH pierde-proteínas se sospechará en niños que, de forma brusca, presentan edemas y en los que se descarta una enfermedad hepática y renal; deberá estudiarse la posibilidad de infección asociada por CMV y/o HP. Aunque no se trate del agente causal, se recomienda efectuar tratamiento erradicador de la infección por HP demostrada, dado que se considera que puede afectar la evolución de la enfermedad. Creemos que la GHPP infantil asociada a infección por CMV debe ser más frecuente de lo que se diagnostica, ya que sólo son evidentes aquellos casos en los que el deterioro clínico provocado por la pérdida excesiva de proteínas origina una clínica que motiva el estudio de estos pacientes.
