


El síndrome Sweet o dermatosis neutrofílica febril fue descrito por primera vez en 1964 y se caracteriza por la tríada de fiebre, lesiones cutáneas eritematosas dolorosas y leucocitosis con neutrofilia1-4. Mientras que en la edad adulta se trata de una entidad bien conocida, sobre todo en mujeres entre 30-50 años1,3, en pediatría son pocos los casos descritos1,2,4. Es conocida su asociación con enfermedades sistémicas, fármacos5 o la gestación1-3, así como con inmunodeficiencias primarias (IDP), entre las que se encuentra la enfermedad granulomatosa crónica (EGC). Se presenta el caso de un adolescente con EGC al que se diagnosticó una variante histiocitoide de esta entidad.
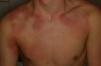
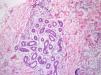
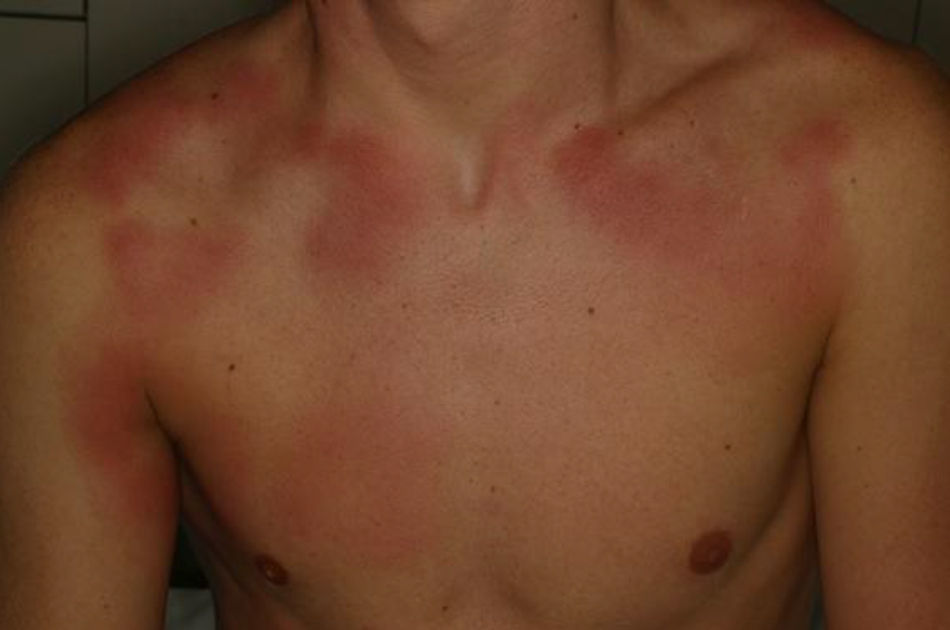
Se trata de un paciente de 15 años afectado de EGC que presentó fiebre de 24 h de evolución y lesiones cutáneas eritematosas, anulares, induradas y dolorosas (fig. 1). No presentaba otro foco de la fiebre y negaba ingesta de sustancias tóxicas, así como de nuevos medicamentos aparte de sus profilaxis habituales con trimetoprim-sulfametoxazol, itraconazol e interferón gamma. El hemograma era normal con una velocidad de sedimentación globular de 18mm/h y una proteína C reactiva de 5,68mg/dL. Se cursaron serologías a Bartonella spp., Borrelia spp., citomegalovirus, Coxiella burnetii, virus de Epstein-Barr, herpesvirus humano 6, Mycoplasma pneumoniae y Toxoplasma gondii, así como un hemocultivo, y se ingresó al paciente con antibioterapia por vía intravenosa (iv). A las 24 h presentó empeoramiento de las lesiones cutáneas y de la fiebre, con afectación del estado general y aparición de leucocitosis con neutrofilia, planteándose el diagnóstico diferencial con entidades inflamatorias-autoinmunes, por lo que se realizó una biopsia cutánea que fue compatible con dermatosis neutrofílica histiocitoide (fig. 2), observándose grupos de linfocitos, histiocitos y células mononucleares en dermis media y profunda. La inmunohistoquímica mostró positividad de las células mononucleares para lisozima, CD163 y CD68 (PGM1) y característicamente para mieloperoxidasa, indicando que se tratara de células mieloides inmaduras. El paciente no cumplía criterios clínicos ni de laboratorio de síndrome hemofagocítico; se decidió iniciar tratamiento corticoideo iv. A las 24 h presentó mejoría de las lesiones y desaparición de la fiebre. Tras obtener resultados microbiológicos negativos, se suspendió la antibioterapia. Posteriormente, se realizó una pauta descendente de corticoides durante 6 semanas con buena evolución. Ante la previsión de un periodo corto de tratamiento corticoideo no se plantearon otras opciones terapéuticas descritas en el paciente inmunodeprimido (gammaglobulina iv) y se decidió cambiar la pauta de profilaxis antifúngica a posaconazol. Revisados los criterios de síndrome de Sweet, se constató el cumplimiento de ambos criterios mayores (inicio abrupto de lesiones cutáneas dolorosas e histología con infiltrado neutrofílico) y todos los menores (enfermedad predisponente, fiebre, aumento de reactantes de fase aguda y excelente respuesta a corticoides).
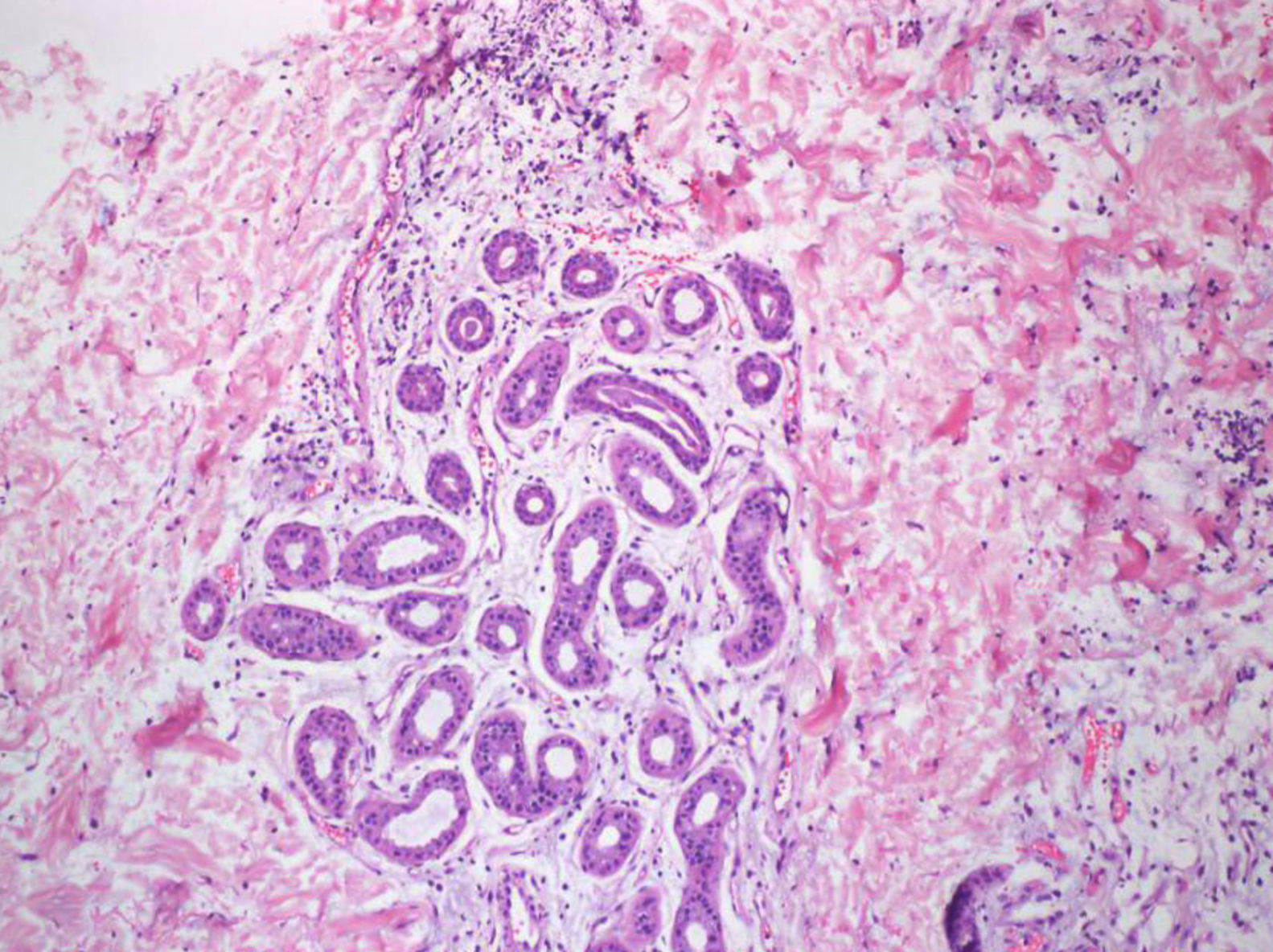
La etiología del síndrome de Sweet es desconocida4. Se implican diferentes mecanismos como una respuesta inespecífica frente virus y bacterias, reacciones de hipersensibilidad y fenómenos de autoinmunidad2. En pediatría, alguna serie describe hasta un 60% de casos asociados a enfermedades sistémicas (reumatológicas, IDP), enfermedades malignas y casos secundarios a fármacos1,2. La histología característica es un infiltrado de neutrófilos maduros situado en la dermis papilar con leucocitoclasia, característicamente sin vasculitis. Sin embargo, en la variante histiocitoide, existe un infiltrado malinterpretado mayoritariamente como de histiocitos, pero que en la inmunohistoquímica presentan inmunorreactividad para mieloperoxidasa, muy indicativo de células mieloides inmaduras6.
La EGC es una IDP en la que se produce un trastorno congénito de la fagocitosis. El defecto bioquímico subyace en la incapacidad de producir especies reactivas de oxígeno por el sistema NADPH oxidasa, condicionando un defecto en la lisis de microorganismos, sobre todo catalasa positivo, generándose una predisposición a infecciones bacterianas y fúngicas3,4,7. Estos pacientes pueden presentar además importantes fenómenos autoinflamatorios3,7, tales como la formación de granulomas, colitis «Crohn-like» y fibrosis pulmonar. Entre los mecanismos descritos relacionados con la hiperinflamación, encontramos una disminución de la apoptosis de los neutrófilos, un disbalance en los receptores de la respuesta inmunitaria innata como los Toll like receptors y los del complemento, una inducción de los linfocitos Th17, un déficit de actividad del Nrf2 y una activación exagerada del inflamosoma. Con la mejoría del tratamiento antibiótico, y sobre todo antifúngico, se ha logrado que muchos pacientes alcancen la edad adulta, apareciendo dichos fenómenos autoinflamatorios con mayor frecuencia y determinando la morbilidad de los mismos8.
Dada la asociación entre síndrome de Sweet y enfermedades sistémicas tales como la EGC, recomendamos considerarla en el diagnóstico diferencial de dicho síndrome. En el sentido opuesto, en todo paciente afectado de EGC que presente un cuadro de fiebre y lesiones cutáneas, no deberíamos olvidar la posibilidad de un fenómeno autoinflamatorio en su diagnóstico diferencial.
