



Sr. Editor:
El feocromocitoma es una de las causas, aunque infrecuente, de hipertensión arterial en niños. En ocasiones, se asocia a alteraciones genéticas, algunas de ellas de reciente descripción. Entre éstas tenemos las del complejo mitocondrial succinato deshidrogenasa (SDH), que se asocian a un mayor grado de malignidad y a la aparición de otro tipo de neoplasia.
Presentamos el caso clínico de un varón de 12 años sin antecedentes personales ni familiares de interés. Fue remitido a nuestro hospital con historia de 1 año de evolución de episodios de cefalea, de localización frontal, pulsátil, de escasas horas de duración, que cedieron con flunarizina (Sibelium®). En la última revisión, se objetivaron cifras de presión arterial por encima del p95 (150/105). El paciente refería en los últimos 4 meses, sudoración excesiva, poliuria, polidipsia, intolerancia al calor y dolor en flanco izquierdo. A la exploración, únicamente, destacaba un peso por debajo del p3. Como exploraciones complementarias de laboratorio, se realizaron hemograma, bioquímica, coagulación, hormonas tiroideas, enolasa neuroespecífica y función renal con resultados normales. Al realizar determinación de catecolaminas en orina de 24 h se obtuvieron los siguientes resultados:
- –
Catecolaminas libres en orina: 1.099 μg/24 h (0–100).
- –
Noradrenalina: 1.068 μg/24 h (32 ± 7).
- –
Adrenalina: 31 μg/24 h (5 ± 2).
- –
Dopamina: 991 μg/24 h (292 ± 56).
Se realizó resonancia magnética (RM) abdominal en la que se halló una masa de 3 × 2 cm en el área paraórtica izquierda, medial al polo inferior del riñón, lobulada y no bien encapsulada. La metayodobencilguanidina (MIBG) fue negativa, probablemente por interferencia del tratamiento antihipertensivo con antagonistas del calcio que estaba recibiendo en ese momento. La ecografía tiroidea, la tomografía computarizada (TC) torácica y la gammagrafía ósea fueron normales.
Con diagnóstico de presunción de feocromocitoma, se inició tratamiento con fenoxibenzamina (la dosis máxima empleada fue de 0,4 mg/kg/día), que se mantuvo durante 21 días, y posteriormente con propranolol por taquicardia (la dosis máxima empleada fue de 0,3 mg/kg/día). Se intervino quirúrgicamente, sin complicaciones, mediante laparotomía, y se realizó extirpación completa de una tumoración de 4 × 3 × 3 cm encapsulada, con peso de 23 g cuyo diagnóstico anatomopatológico fue de paraganglioma. A los 15 días de la intervención, el paciente se encontraba asintomático con cifras de presión arterial y catecolaminas en orina normales. El estudio genético demostró un cariotipo normal, y se halló una mutación en el gen succinato deshidrogenasa subunidad B (SDHB), c166-170 del CCTCE. Posteriormente, se realizó estudio genético-molecular familiar en el que se halló que tanto el padre como dos tíos paternos eran portadores de la misma mutación (fig. 1).
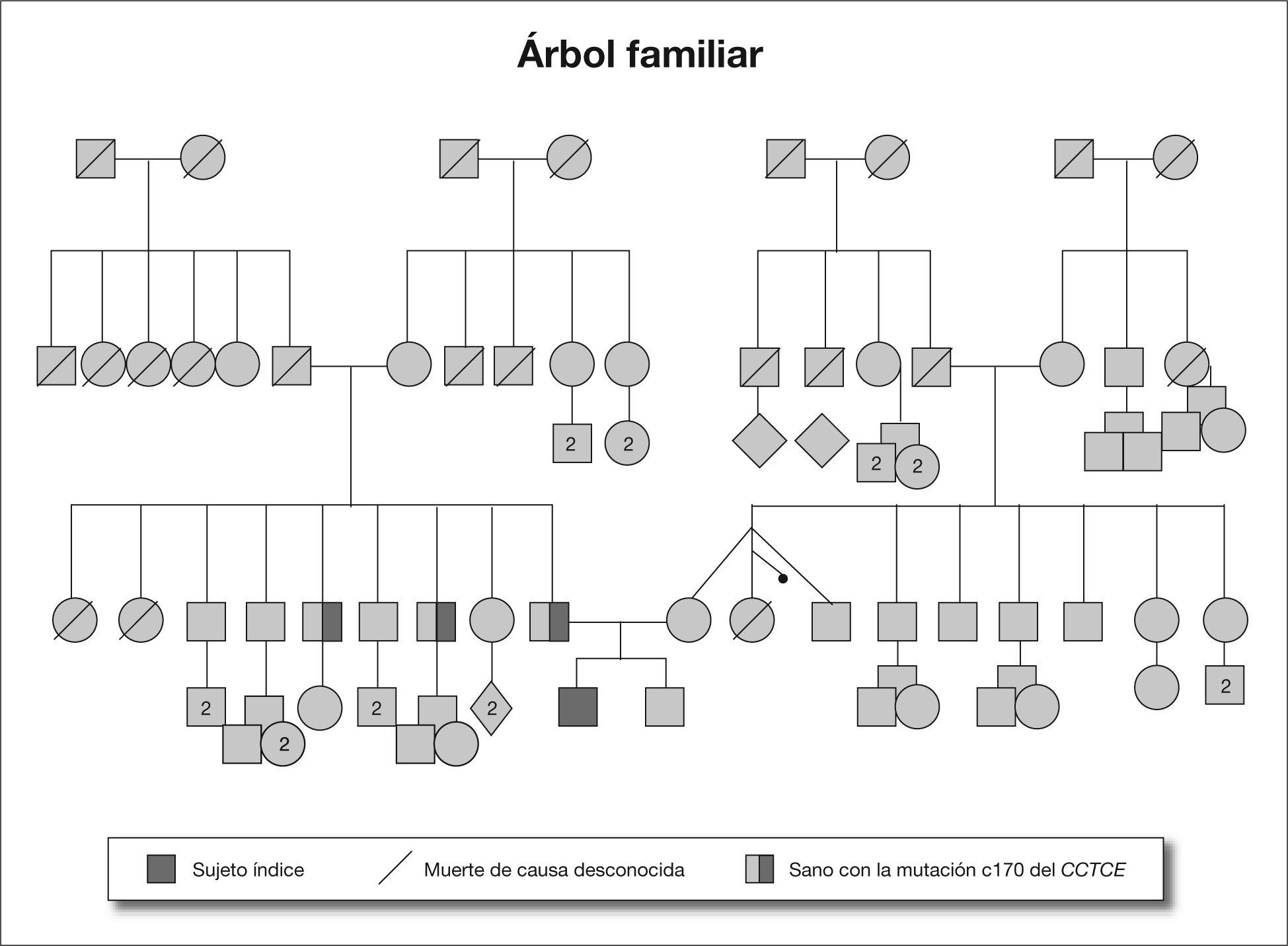
El feocromocitoma es un tumor poco frecuente en la edad pediátrica, derivado de las células cromafines de la cresta neural, de localización, en la glándula suprarrenal o extraadrenal en los ganglios autónomos de la cadena simpática paravertebral. Su máxima incidencia se sitúa entre los 8 y los 14 años.
En la infancia, a diferencia de la edad adulta, el porcentaje de feocromocitomas familiares y de localización extraadrenal es mayor (alrededor de un 30 %). La forma de presentación más frecuente sigue siendo la forma aislada (70-80 %) y de localización suprarrenal1,2.
Las manifestaciones clínicas del feocromocitoma son consecuencia en su mayoría de la producción elevada de catecolaminas. Las más características son hipertensión arterial, cefalea y diaforesis1,2.
El tratamiento es quirúrgico, tras la localización del tumor mediante distintas pruebas de imagen, y un adecuado bloqueo alfa, preoperatorio, que reduzca los riesgos asociados al procedimiento.
Una presentación a edad temprana y de localización extraadrenal debe hacernos sospechar una alteración genética. Por este motivo, debe realizarse estudio genético-molecular en busca de algunos de los síndromes en los que se ha demostrado una susceptibilidad para el desarrollo de feocromocitoma como son: MEN tipo 2, el síndrome de Von Hippel Lindau, neurofibromatosis tipo 1 y, recientemente, las mutaciones en los genes que codifican tres de las cuatro subunidades del complejo mitocondrial SDH denominado síndrome paraganglionar familiar (PGL)2–6.
Las subunidades del complejo SDH implicadas en el desarrollo de paragangliomas son la B (PGL4), C (PGL3) y D (PGL1). La mutación en la subunidad B está relacionada con un mayor grado de malignidad, su presentación a edades tempranas y con una mayor predisposición para desarrollar carcinoma de células renales.
La presencia de una mutación en el gen SDHB, como ocurre en nuestro caso, debe ser considerada un factor de riesgo de malignidad y recurrencia, que nos exige un estricto seguimiento del paciente (historia clínica, exploración física, catecolaminas en orina de 24 h y pruebas de imagen como TC/RM ± MBIG o dopamina-PET)3 y de los casos positivos para la mutación en el resto de la familia2–6.
