










El cribado neonatal de la aciduria glutárica tipo 1(AG-1) ha supuesto un cambio radical en la evolución y el comienzo de la enfermedad. En este estudio se analizan los resultados de los primeros 5 años (2015-2019) del programa de cribado neonatal (PCN) de AG-1 en Asturias.
MaterialSe diseñó un estudio observacional, descriptivo y retrospectivo. La población estaba formada por neonatos participantes en el PCN nacidos entre el 1 de enero de 2015 y el 31 de diciembre de 2019. La determinación de glutarilcarnitina (C5DC) en sangre impregnada en papel se realizó mediante espectrometría de masas en tándem con punto de corte en 0,25μmol/l.
ResultadosEl cribado se llevó a cabo en 30.120 recién nacidos. En el análisis del marcador C5DC se encontraron diferencias en la concentración dependiendo de la edad gestacional, del tipo de alimentación y de las horas de vida a la extracción. Estas diferencias no fueron relevantes a efectos del cribado. No se observaron diferencias entre menores y mayores de 1.500g. Se identificaron 2 casos afectos y 3 falsos positivos. No se detectaron falsos negativos. Los diagnósticos fueron confirmados mediante estudio genético. Los pacientes reciben tratamiento desde el diagnóstico y no han presentado crisis encefalopáticas los primeros 4 años de vida.
ConclusionesEl cribado ha permitido el diagnóstico precoz de 2 casos de AG-1 en los 5 primeros años desde su instauración en nuestra comunidad. Aunque existen diferencias en los niveles de C5DC en función de la edad gestacional, del tipo de alimentación y de las horas de vida a la extracción, estas no tuvieron trascendencia para el cribado.
Neonatal screening of glutaric aciduria type 1 (GA-1) has brought radical changes in the course and outcomes of this disease. This study analyses the outcomes of the first 5 years (2015-2019) of the AGA1 neonatal screening programme in our autonomous community.
MaterialWe conducted an observational, descriptive and retrospective study. All neonates born between January 1, 2015 and December 31, 2019 that participated in the neonatal screening programme were included in the study. The glutarylcarnitine (C5DC) concentration in dry blood spot samples was measured by means of tandem mass spectrometry applying a cut-off point of 0.25μmol/l.
ResultsA total of 30 120 newborns underwent screening. We found differences in the C5DC concentration based on gestational age, type of feeding and hours of life at sample collection. These differences were not relevant for screening purposes. There were no differences between neonates with weights smaller and greater than 1500g. Screening identified 2 affected patients and there were 3 false positives. There were no false negatives. The diagnosis was confirmed by genetic testing. Patients have been in treatment since diagnosis and have not developed encephalopathic crises in the first 4 years of life.
ConclusionsScreening allowed early diagnosis of two cases of GA-1 in the first 5 years since its introduction in our autonomous community. Although there were differences in C5DC levels based on gestational age, type of feeding and hours of life at blood extraction, they were not relevant for screening.

La aciduria glutárica tipo 1 (AG-1) es un error innato del metabolismo, de herencia autosómica recesiva, que tiene su origen en el déficit de la glutaril-CoA deshidrogenasa (GCDH), enzima que lleva a cabo la deshidrogenación y decarboxilación de glutaril-CoA en la vía metabólica de degradación de la lisina, hidroxilisina y triptófano. El bloqueo de dicha vía produce un aumento de glutaril-CoA, glutaconil-CoA y secundariamente acumulación de ácido glutárico y 3-hidroxiglutárico, metabolitos con efectos neurotóxicos1. Los pacientes no detectados precozmente presentan una clínica predominantemente neurológica, con hipotonía, retraso psicomotor, temblores y distonías2. Un signo característico es la macrocefalia que destaca a partir de los 6 meses, aunque puede ser detectada intraútero3. El comienzo hasta en un 75% de los casos es agudo presentándose en forma de crisis encefalopática. Esta manifestación consistente en la disminución del nivel de consciencia, convulsiones, irritabilidad y la aparición de signos extrapiramidales, suele aparecer en los 6 primeros años de vida1,4. En los estudios de neuroimagen se pueden observar hallazgos como hipoplasia fronto-opérculo-temporal, destrucción de ganglios de la base, dilatación quística de la cisura de Silvio, leucoencefalopatía o hematomas subdurales; siendo incluido en ocasiones como parte del diagnóstico diferencial del síndrome del niño zarandeado5.
En el cribado de la AG-1 se lleva a cabo mediante la determinación de glutarilcarnitina (C5DC) en sangre impregnada en papel mediante espectrometría de masas en tándem (MS/MS). En Asturias esta detección se incorporó al programa de cribado neonatal (PCN) a finales del año 20146,7.
En los últimos años se ha estudiado la capacidad de detección del marcador C5DC, especialmente en pacientes bajo excretores que pueden presentar niveles de C5DC dentro de la normalidad.
El objetivo principal de este estudio fue analizar los resultados de los primeros cinco años (2015-2019) de cribado neonatal de AG-1.
MaterialSe diseñó un estudio observacional, descriptivo y retrospectivo. La población a estudio estaba formada por los participantes en el PCN, todos ellos nacidos entre el 1 de enero de 2015 y el 31 de diciembre de 2019. Los datos fueron obtenidos del registro del laboratorio de cribado neonatal, ubicado en el servicio de bioquímica clínica del Hospital Universitario Central de Asturias (HUCA) desde 2014, que incluye todos los cribados metabólicos realizados en los hospitales públicos y privados de la comunidad autónoma.
De cada paciente se recogió fecha de nacimiento, hospital en el que se tomaba la muestra, edad gestacional (EG), peso al nacimiento, tipo de alimentación, horas de vida a la extracción de la muestra y concentración de C5DC (μmol/l).
El estudio recibió la autorización del Comité de Ética de la Investigación (Código 2021.141).
La muestra utilizada para el cribado fue sangre capilar obtenida tras la punción del talón e impregnada en papel Whatman® 903. Para su obtención se estableció un protocolo de recogida entre las 48 y 72h de vida, mediante un dispositivo automático de incisión o lanceta estéril. La tarjeta de filtro se debía impregnar adecuadamente con una gota de sangre y cumplimentar con los datos del paciente. La muestra se identificó mediante un código de barras y un número de referencia. Posteriormente, se procedió al envío al laboratorio a través de circuitos intra e interhospitalarios específicos7.
El procedimiento analítico para la determinación de C5DC se realizó mediante MS/MS, previa ionización con electrospray (ESI). El análisis se realizó en espectrómetro de masas triple cuadrupolo (QTRAP® 5500; AB Sciex) acoplado a un cromatógrafo de líquidos (Ekspert® Ultra LC 100; Eksigent).
El punto de corte establecido para el marcador C5DC durante los 5 años se mantuvo constante en 0,25μmol/l. Este valor se ajustó tras analizar los resultados obtenidos en un primer pilotaje en 2014 en el que se había fijado como punto de corte 0,17μmol/l y se validó anualmente mediante la revisión periódica de los percentiles.
Se realizó un análisis descriptivo proporcionando distribuciones de frecuencias absolutas y relativas para variables cualitativas, y medidas de posición y dispersión en el caso de variables cuantitativas. Las diferencias de variables cuantitativas entre más de 2 grupos se evaluaron mediante el test Kruskal-Wallis ante el incumplimiento de las hipótesis de normalidad y/o homocedasticidad. Cuando las diferencias alcanzaron significación estadística se realizó el test pos hoc de Dunn. Para el estudio comparativo de variables entre 2 grupos, el test que se empleó fue t de Student, con la corrección de Welch ante varianzas diferentes. El nivel de significación fijado fue de 0,05. El análisis estadístico se efectuó con los programas IBM SPSS® Statistics 2023 (IBM Corporation, Armonk New York, EE. UU.) y R Development Core Team (R Foundation for Statistical Computing, Vienna, Austria), versión 4.1.3R (2022).
Para conocer la cobertura poblacional del PCN se obtuvieron los datos de recién nacidos (RN) registrados en la página del Instituto Nacional de Estadística (INE)8.
Se usaron los puntos de corte de la Organización Mundial de la Salud (OMS) para la categorización de la EG en los siguientes grupos: prematuros extremos (EG<28 semanas), muy prematuros (≥28 y<32), moderados (≥32 y <34), tardíos (≥34 y <37) y RN a término (≥37)9.
Se definió como bajo peso para la edad gestacional (BPEG) a los RN de ambos sexos con un peso medio por debajo del p10 obtenido de las gráficas de Fenton 201310.
ResultadosEl cribado de AG-1 se llevó a cabo en 30.120RN en nuestra comunidad entre 2015 y 2019 (fig. 1). La cobertura del cribado fue superior al 99,6% en todos los años. En el año 2019 fueron cribados todos los RN de Asturias y 2RN residentes en otras comunidades autónomas, alcanzando una cobertura del 100%.

En la muestra se obtuvo un valor para el p99 de C5DC de 0,24μmol/l, p99,5 de 0,26μmol/l y p99,9 de 0,33μmol/l. Tras la primera determinación de C5DC, 216 muestras de pacientes superaron el valor del punto de corte (0,25μmol/l). En una segunda determinación 5 continuaron manteniéndose por encima, siendo finalmente confirmado el diagnóstico en 2. Ambos casos eran RN a término, con peso adecuado para la edad gestacional (PAEG) y asintomáticos. Uno recibía lactancia materna (LM) y otra lactancia mixta (L mixta). Durante los 5 años de cribado, se identificaron 3 casos falsos positivos (FP) y 2 verdaderos positivos (VP), descritos en la tabla 1. Hasta el momento no han sido detectados falsos negativos (FN). La incidencia de AG-1 fue de uno por 15.060RN/año.
Descripción de los pacientes con cribado positivo de aciduria glutárica tipo 1
| Pacientes positivos PCN | Caso 1 | Caso 2 | FP1 | FP2 | FP3 |
|---|---|---|---|---|---|
| Diagnóstico | 12ddv | 13 ddv | — | — | — |
| C5DC (μmol/l) | 8,26 | 6,04 | 0,94 | 0,31 | 0,37 |
| Clínica al nacimiento | — | — | — | Distrés respiratorio | — |
| Hipoglucemia neonatal | |||||
| Alimentación | LM | L mixta | LM | LA | L mixta |
| Peso | PAEG | PAEG | PAEG | BPEG | PAEG |
| EG | 39 | 39 | 38 | 38 | 40 |
C5DC: glutarilcarnitina; ddv: días de vida; EG: edad gestacional; FP: falsos positivos; L mixta: lactancia mixta; LA: lactancia artificial; LM: lactancia materna; PAEG: peso adecuado para la edad gestacional; PCN: programa de cribado neonatal.
Se realizó estudio genético a ambos pacientes siendo uno portador heterocigoto de las variantes c.374T>C (p.Leu125Pro) y c.1157G>C (p.Arg386Pro) en el gen GCDH, ambas considerados de significado incierto; y el otro caso doble portador heterocigoto de las variantes patogénicas c.877G>A (p.Ala293Thr) y c.1204C>T (p.Arg402Trp) compatibles con AG-111.
Desde el nacimiento hasta la actualidad los 2 pacientes presentan un desarrollo psicomotor normal habiendo cumplido los primeros 4 años de vida sin presentar crisis encefalopáticas.
Se disponía de datos de edad gestacional de 30.106RN (99,95%). El 93,3% de los nacimientos totales fueron a término y el 6,7% prematuros con la distribución que se muestra en la figura 2.
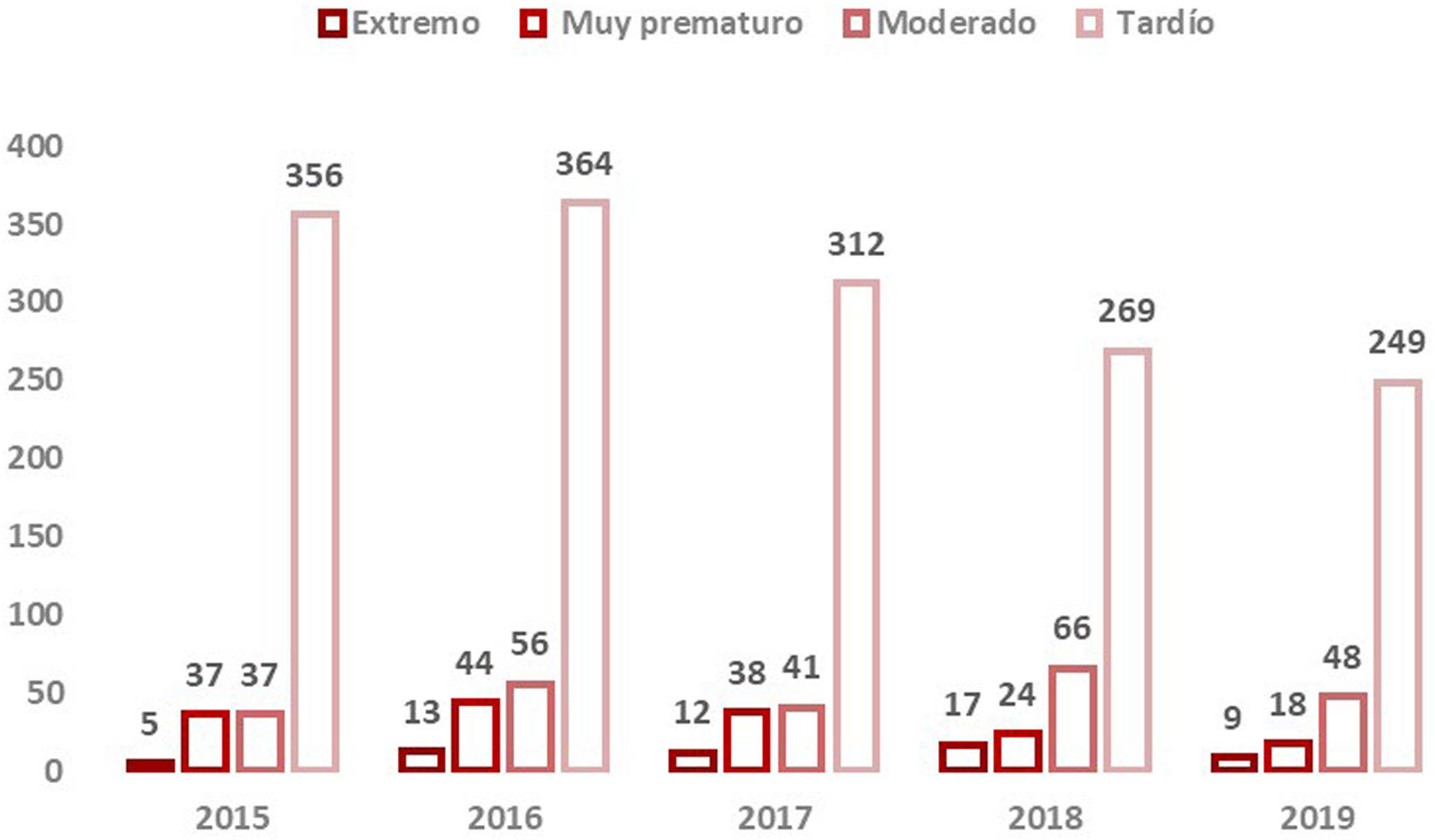
En el análisis se objetivaron diferencias en la concentración de C5DC según la edad gestacional (p<0,001). En el análisis por grupos de prematuros estas diferencias se observaron entre RN a término y prematuros tardíos, a término y prematuros moderados, a término y muy prematuros, prematuros tardíos y prematuros moderados, y prematuros moderados y prematuros extremos (tabla 2).
Resultado del análisis estadístico (p-valor) al comparar los valores de C5DC según la edad gestacional
| C5DCp25-p75 (μmol/l) | A término (0,12-0,15) | Prematuro tardío (0,1-0,15) | Prematuro moderado (0,09-0,14) | Muy prematuro (0,09-0,15) | Prematuro extremo (0,11-0,17) |
|---|---|---|---|---|---|
| A término (0,12-0,15) | — | <0,001 | <0,001 | 0,004 | 0,322 |
| Prematuro tardío (0,1-0,15) | <0,001 | — | 0,003 | 0,367 | 0,072 |
| Prematuro moderado (0,09-0,14) | <0,001 | 0,003 | — | 0,301 | 0,001 |
| Muy prematuro (0,09-0,15) | 0,004 | 0,367 | 0,301 | — | 0,053 |
| Prematuro extremo (0,11-0,17) | 0,322 | 0,072 | 0,001 | 0,053 | — |
C5DC: glutarilcarnitina.
Entre los grupos de neonatos a término y prematuros tardíos, a término y prematuros moderados, a término y muy prematuros, prematuros tardíos y prematuros moderados, y prematuros moderados y prematuros extremos existían diferencias (p valor <0,05).
En negrita, los valores estadísticamente significativos (< 0,05).
El peso medio al nacimiento fue de 3.211g con p50 de 2.240g. El peso mínimo fue 500g y el máximo 5.960g. El 0,8% (235) de los RN pesaron menos de 1.500g. Se cuantificaron el 12,6% de RN con BPEG. Al comparar la concentración de C5DC según el peso fuera menor o mayor de 1.500g, no se observaron diferencias estadísticamente significativas (tabla 3). Sin embargo, si existían diferencias cuando se compararon RN con BPEG frente a RN con peso adecuado para la edad gestacional (PAEG).
Valores de C5DC (p25-p75) en función del peso al nacimiento en neonatos con bajo peso para la edad gestacional (BPEG), peso adecuado (PAEG), <1.500 y ≥1.500g. Se encontraron diferencias (p-valor) entre BPEG y PAEG
| BPEG | PAEG | p-valor | |
|---|---|---|---|
| C5DCp25-p75(μmol/l) | 0,1-0,15 | 0,11-0,15 | <0,001 |
| <1.500g | ≥1.500g | p-valor | |
|---|---|---|---|
| C5DCp25-p75(μmol/l) | 0,1-0,16 | 0,11-0,15 | 0,7 |
BPEG: bajo peso para la edad gestacional; C5DC: glutarilcarnitina; PAEG: peso adecuado para la edad gestacional.
El tipo de alimentación más frecuente fue la alimentación materna (57,3%), seguida de la lactancia artificial (LA) (21,1%) y L mixta (20,9%), 101 (0,4%) muestras pertenecían a RN que habían recibido nutrición parenteral (NP). En el análisis se objetivaron diferencias en el valor de C5DC según el tipo de alimentación. Al comparar la concentración de C5DC en los distintos grupos, estas diferencias se observaron entre alimentados con LM y LA, LM y L mixta, LA y L mixta y, NP y LM (tabla 4).
Resultado del análisis estadístico (p-valor) al comparar los valores de C5DC según el tipo de alimentación
| C5DCp25-p75 (μmol/l) | LM (0,11-0,16) | LA (0,1-0,14) | L mixta (0,1-0,15) | NP (0,1-0,14) |
|---|---|---|---|---|
| LM (0,11-0,16) | — | <0,001 | <0,001 | <0,001 |
| LA (0,1-0,14) | <0,001 | — | <0,001 | 1 |
| L mixta (0,1-0,15) | <0,001 | <0,001 | — | 0,535 |
| NP (0,1-0,14) | <0,001 | 1 | 0,535 | — |
C5DC: glutarilcarnitina; L mixta: lactancia mixta; LA: lactancia artificial; LM: lactancia materna; NP: nutrición parenteral.
En negrita, los valores estadísticamente significativos (< 0,05).
La edad mediana de toma de la muestra de sangre capilar fue las 52h. La mayoría de extracciones se llevaron a cabo a las 48h de vida. Al estudiar el comportamiento de C5DC según la edad a la que se tomó la muestra, se encontraron diferencias entre la concentración de C5DC de RN a los que se les tomó la muestra con más de 72h de vida respecto a los que tenían entre 48 y 72h y los que tenía menos de 48h de vida (tabla 5).
Resultado del análisis estadístico (p-valor) al comparar los valores de C5DC según horas de vida a las que se tomó la muestra para el cribado
| C5DC p25-p75 (μmol/l) | <48 hdv (0,11-0,16) | 48-72hdv (0,11-0,15) | >72hdv (0,11-0,14) |
|---|---|---|---|
| <48hdv (0,11-0,16) | — | <0,424 | <0,001 |
| 48-72hdv (0,11-0,15) | <0,424 | — | <0,001 |
| >72hdv (0,1-0,14) | <0,001 | <0,001 | — |
C5DC: glutarilcarnitina; hdv: horas de vida.
En negrita, los valores estadísticamente significativos (< 0,05).
La AG-1 fue incorporada al PCN en nuestra comunidad a finales del año 2014 conforme a la resolución de la Dirección General de Salud Pública publicada en el Boletín Oficial del Estado (BOE) el 6 de noviembre de 2014.
En estos 5 primeros años de cribado se detectaron y confirmaron 2 casos de AG-1. Son reseñables la alta cobertura poblacional del PCN y el descenso progresivo y preocupante de la natalidad.
La incidencia de AG-1 resulta mayor a la estimada actualmente en Europa (1/120.000RN). En el año 2021 se publicó en la Revista Española de Salud Pública un estudio que evaluaba la prevalencia global de las enfermedades metabólicas partiendo de los informes publicados por la Asociación Española de Cribado Neonatal (AECNE) y el Ministerio de Sanidad entre 2016 y 20186,12. La AG-1 se diagnosticó mediante cribado en todas las comunidades menos en Aragón, sumando 57 casos y siendo las comunidades con mayor prevalencia Andalucía, Madrid y Galicia (inferior a 1:55.000RN).
En la Comunidad de Madrid entre los años 2011 y 2019 se diagnosticaron mediante cribado 12 casos de AG1, estando todos asintomáticos al diagnóstico. Se estimó una incidencia de 1:49.402. No fueron detectaron falsos negativos13. En Galicia durante 20 años de cribado (2000-2019), se detectaron 7 casos de AG1 y se diagnosticó un bajo excretor que había resultado negativo (FN). Se estimó una incidencia de 1:57.80214. La mayoría de las comunidades en este periodo establecieron su punto de corte ≥p99,5.
En nuestra comunidad no han sido detectados FN. Los 3 casos identificados como FP se correspondían con RN sanos, a término, que recibían distintos tipos de alimentación enteral (LM, LA y L mixta) y uno de ellos fue BPEG. En una serie de casos publicada recientemente se asoció la insuficiencia renal a la elevación de C5DC en RN, si bien, ningún FP de este estudio presentaba tal afectación15. Se debe tener en cuenta que el marcador C5DC presenta la misma relación m/z (masa-carga) que 3-OH hexanoilcarnitina (C6-OH). En la literatura se recogen FN en muestras de RN mayores de 7 días de vida bajos excretores con niveles de C5DC normales. Aunque también otras series de casos publicadas no objetivan diferencias en los valores de C5DC en sangre entre pacientes altos y bajos excretores16,17.
En cuanto al estudio genético, las variantes A293T y R402W encontradas se corresponden con las más frecuentes en nuestro país según las series publicadas13,14,18. R402W también es la mutación más común en Portugal e Italia19. En cambio, las variantes de significado incierto (L125P y R386P) no han sido descritas en nuestra región previamente.
En nuestro estudio parecen existir diferencias en los niveles de C5DC según EG, tipo de alimentación y hora de extracción de la muestra, aunque las mismas no tienen relevancia en los resultados del cribado. Un estudio americano relacionó el nivel socio económico y el BPEG de manera independiente y conjunta con los metabolitos del cribado, viéndose interacción entre estas variables y el marcador C5DC20.
La evolución clínica de los pacientes afectos de AG-I ha cambiado radicalmente desde el diagnóstico a través del PCN21,22. Cuando los pacientes afectos no eran detectados y tratados precozmente, el 90% comenzaba con una crisis encefalopática aguda con afectación motora. El deterioro neurológico que supone la enfermedad es progresivo e irreversible, y conlleva a la atrofia cerebral y el deterioro cognitivo. El tratamiento está basado en una dieta restringida en lisina. Para ello, se utilizan fórmulas de aminoácidos exentas en lisina, reducidas en triptófano y enriquecidas en arginina. La suplementación con carnitina está indicada de por vida para disminuir el estrés oxidativo y las posibles complicaciones cerebrales secundarias a su agotamiento23.
En los estudios de comparación se objetiva que el 90% de pacientes que inician el tratamiento en el periodo neonatal permanece asintomático y las crisis encefalopáticas disminuyen al 10-20% de los casos sintomáticos24. Además, se ha observado una reducción de la afectación de la deglución, bipedestación y distonías del 75%25. Los casos de este estudio no han desarrollado crisis hasta la fecha.
Como fortalezas del trabajo, destacar que este es el primer estudio que expone los datos del cribado de AG-1 en nuestra comunidad desde su implantación, que además ha permitido detectar en esta comunidad autónoma dos casos de una enfermedad rara. Además, los marcadores de este PCN están acreditados por norma UNE EN-ISO 15189, dotándolo de herramientas para el aseguramiento de la calidad.
La principal limitación de este estudio es el tiempo, ya que se ha centrado en 5 años (2015-2019) debido a la necesidad de disponer de un registro de datos de años completos. En febrero de 2020 se llevó a cabo un cambio de proveedor del reactivo optándose por evaluar este primer periodo para no introducir sesgos.
Este trabajo asienta las bases para una comparación con el periodo de 5 años siguiente (2020-2024), donde se podrá valorar con mucha mayor experiencia y casuística el algoritmo de cribado, así como la pertinencia de implementar otras posibles mejoras, como es la inclusión de segundos marcadores.
ConclusiónEl diagnóstico precoz de AG1 mediante el cribado neonatal de C5DC es clave para modificar la evolución natural de la enfermedad y evitar el daño neurológico en los pacientes. El PCN ha permitido el diagnóstico precoz de dos casos de AG-1 en los cinco primeros años desde su instauración, sin tener constancia de FN hasta el momento.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
