
Introducción
La enfermedad de células falciformes es una alteración genética de la hemoglobina. Su herencia es autosómico recesiva, distinguiéndose a los portadores heterocigotos de una sola mutación en el locus de la globina (resultando la llamada hemoglobina S), los cuales permanecerán sanos durante toda su vida, y a los homocigotos o dobles heterocigotos, que padecerán la sintomatología característica de la enfermedad1: anemia hemolítica crónica, crisis dolorosas por oclusión de vasos y elevado riesgo infeccioso por asplenia funcional.
Varios países en Europa y América del Norte han incluido la detección de hemoglobinopatías estructurales en los programas de cribado neonatal en los últimos años1-3, teniendo en cuenta que las complicaciones clínicas de los pacientes con enfermedad falciforme son frecuentes, sobre todo en los primeros 3 años de vida4. Diversos estudios que comunican casos de enfermedades infecciosas graves como sepsis, neumonías o meningitis, secuestros esplénicos o crisis de dolor infradiagnosticadas por desconocimiento de la patología de base5, hicieron concluir que era conveniente una detección precoz. Las publicaciones que demuestran los beneficios de una pronta identificación de los niños con enfermedad falciforme frente a un diagnóstico tardío cuando ya ha aparecido una complicación son muy escasas6-8, y no existe ningún ensayo clínico al respecto. Sin embargo, un ensayo aleatorizado, doble ciego, con control con placebo, demostró una dramática disminución en la incidencia de bacteriemia por Streptococcus pneumoniae en niños menores de 3 años con anemia falciforme tratados con profilaxis con penicilina9. En España, la anemia falciforme era una enfermedad rara hasta hace unos 10 años, cuando los pediatras empezaron a familiarizarse con esta enfermedad emergente debido al aumento en la inmigración. La Sociedad Española de Hematología Pediátrica (SEHP)10 detectó con prontitud el aumento en la incidencia y nombró un comité que diseñó un protocolo de diagnóstico, seguimiento y tratamiento en el año 200010. Hasta el año 2004, se registraron un total de 138 pacientes en todo el país seguidos en 24 hospitales, aunque seguramente estas cifras infraestiman la situación real11.
Aunque se han realizado estudios parciales sobre la incidencia de la enfermedad en nuestro medio, la implantación de un programa sistemático de cribado no ha sido aprobada por las autoridades sanitarias hasta recientemente. La Comunidad de Madrid (CM) a instancias de su Instituto de Salud Pública, fue la primera comunidad del país en iniciar el desarrollo del cribado neonatal universal de hemoglobinopatías en mayo de 2003. Presentamos aquí los resultados de los primeros 32 meses de este programa.
Material y métodos
La población diana incluyó todo recién nacido en cualquier centro público o privado de la CM desde mayo de 2003 a diciembre de 2005. La muestra de sangre capilar en papel se obtuvo de la misma tarjeta de la prueba del talón utilizada en el programa de detección precoz de hipotiroidismo congénito e hiperplasia suprarrenal congénita, que se obtiene en las maternidades a partir de las 48 h de vida, antes del alta del niño. Esta muestra se traslada diariamente por mensajería desde todos los hospitales públicos y privados, hasta el laboratorio único responsable del procesamiento de todas las primeras muestras del talón en la CM (Laboratorio de Cribado Neonatal de Enfermedades Endocrino Metabólicas del Hospital Universitario Gregorio Marañón).
El método usado para el análisis de variantes de hemoglobina fue la cromatografía líquida de alta resolución (HPLC), capaz de detectar hemoglobina (Hb) F, A, S, C, D y E en un tiempo de elución de 3 min. El procedimiento analítico utilizado (Variant-BioRad®), permite la automatización y el análisis de cientos de muestras diarias con una alta sensibilidad y especificidad12. Los resultados positivos se repitieron en el mismo papel de filtro, y se confirmaron con una segunda muestra del niño obtenida al citarle en la consulta. Las variantes de cadena gamma desaparecieron o tuvieron una disminución muy marcada en la repetición. Los valores altos de HbA se trataron como sospechosos de haber recibido transfusión previa, y se ofreció repetir más adelante.
Se comunicó a los padres la totalidad de las variantes de hemoglobina, incluyendo los heterocigotos y las hemoglobinas no caracterizadas. En la entrevista con las familias, y tras firma de consentimiento informado, se obtuvo como método confirmatorio una segunda muestra de sangre capilar del talón impregnada sobre papel absorbente, analizado igual que el primero o con un gradiente más expandido si estaba indicado, y ampliando a análisis de ADN si era necesario. El estudio del resto de miembros de la familia se realizó según el deseo de los padres, pero nuestro grupo siempre recomendó el análisis de ambos progenitores y de todos los hermanos.
Los niños con enfermedad falciforme homocigotos para HbS o heterocigotos compuestos iniciaron desde los 2 meses profilaxis con penicilina y vacunación según el protocolo nacional de la SEHP10, y se programó la educación sanitaria. Todos ellos fueron citados en las consultas de seguimiento de esta patología de sus hospitales de referencia cercanos al domicilio respectivo.
Los lactantes con rasgo falciforme (portadores heterocigotos de HbS o C), o heterocigotos para HbD o E, fueron informados de la naturaleza benigna de esta condición, aportándoles información escrita sobre las recomendaciones de la SEHP y asegurando el consejo genético.
Con el fin de evitar falsos positivos, se realizó una electroforesis convencional a los 3 meses de edad en enfermedad falciforme, y a los 12 meses en portadores, ampliando a análisis más sofisticados si estaba indicado.
Resultados
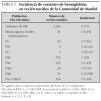
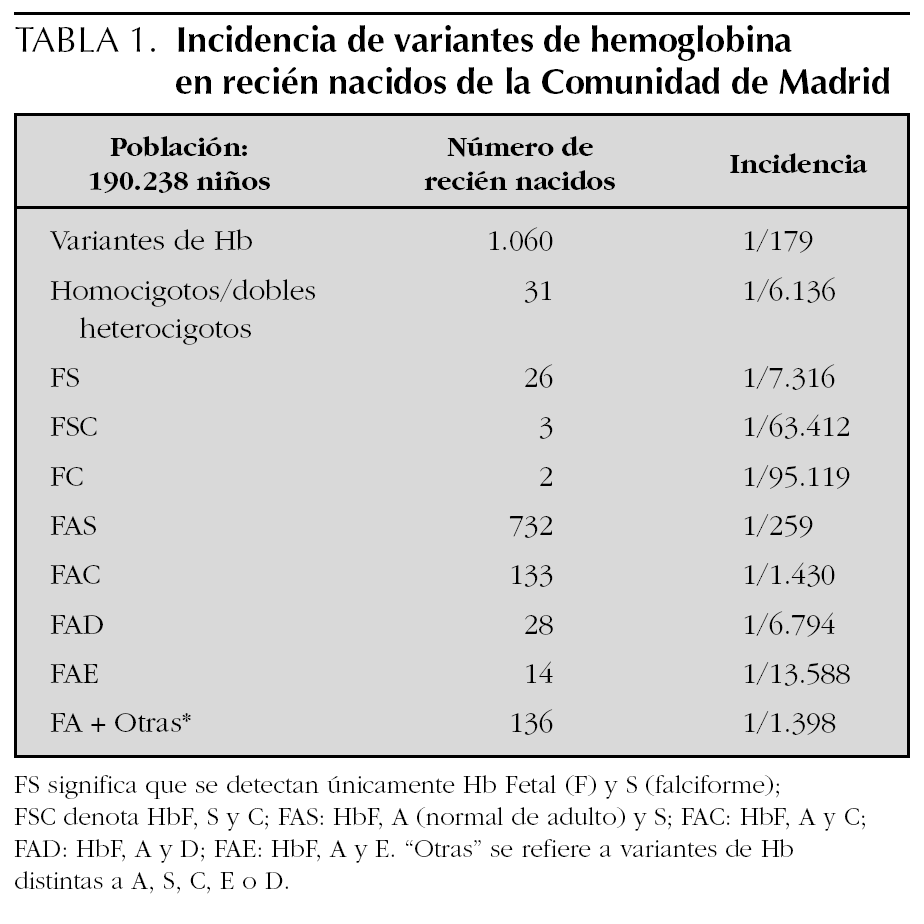
Se analizaron 190.238 recién nacidos, encontrándose 1.060 variantes de hemoglobina (5,59/1.000) durante los primeros 3 años naturales (32 meses) del programa de cribado neonatal de hemoglobinopatías de la Comunidad de Madrid. Un total de 31 de ellos padecían enfermedad grave (homocigotos o dobles heterocigotos) y el resto eran portadores. En la tabla 1 se presentan los patrones de hemoglobina encontrados.
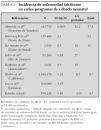
De todas las alteraciones detectadas, se consiguió contacto telefónico en 1.007 familias y finalmente acudieron a consulta 975 (92 %). La edad media en el momento de la primera exploración fue de 39 días (intervalo 8 a 218). Sólo un paciente de los considerados graves (31 pacientes) no pudo ser localizado; la exploración física de éstos fue normal, salvo en un caso que presentaba una marcada ictericia secundaria a una colestasis neonatal no relacionada con la enfermedad falciforme. La relación niño/niña fue 1. El peso medio de recién nacido fue de 3.189 g (intervalo de 606 a 5.260) y 39 semanas de edad gestacional (24 a 42).
Se estudió a la madre en 921 casos, al padre en 880, y se encontró a un total de 102 hermanos con alguna alteración estructural de Hb. Entre los heterocigotos, se identificó herencia paterna en 374 niños (35 %) y materna en 263 (25 %). Un total de 23 neonatos que eran sólo portadores tenían ambos padres con rasgo falciforme, lo que ha motivado con posterioridad la realización de 3 diagnósticos prenatales por biopsia de corion: el resultado fue de 2 fetos sin enfermedad y un portador de rasgo falciforme. Un 11 % de los pacientes no tenía herencia materna de la variante de Hb, pero el padre no fue estudiado. Un total de 5 niños portadores de rasgo falciforme y uno de HbE no tenían evidencia de alteración de la Hb en ninguno de los "declarados" progenitores. El resto de herencias no pudieron asignarse porque ninguno de los progenitores se realizó el estudio, o bien porque la madre era portadora y no se tuvieron datos del padre.
Los padres portadores pertenecían a 44 países distintos. Considerando sólo el rasgo con HbS parental, las naciones más prevalentes de origen fueron Nigeria (18 %), República Dominicana (16 %), Ecuador (9,5 %), España (8 %), Guinea Ecuatorial (6 %), Colombia (5 %) y Marruecos (4 %). Sólo 6 de los progenitores tenían enfermedad falciforme: 3 homocigotos SS, uno doble heterocigoto SC y 2 falciforme-talasemia SB+. Se han producido 2 fallecimientos no relacionados con la alteración de la Hb sino con patología perinatal: uno por prematuridad en un homocigoto SS (24 semanas de edad gestacional) y otro por sepsis neonatal precoz en un niño con rasgo falciforme.
No fue detectado ningún falso positivo, considerando éste como discordancia entre el análisis extraído al nacimiento en la maternidad y el de la segunda muestra obtenida en la consulta de un determinado lactante identificado por la primera prueba. Los profesionales implicados en el programa no han tenido noticia de ningún caso de enfermedad falciforme nacido en Madrid y con resultado del cribado informado como normal, aunque obviamente no se puede evaluar la existencia de falsos negativos.
Discusión
La implantación del cribado neonatal universal de hemoglobinopatías estructurales en la CM se ha beneficiado del complejo entramado previamente establecido para otras enfermedades: El hipotiroidismo congénito y la hiperplasia suprarrenal se analizan en la primera prueba del talón desde 1973 y 1990 respectivamente, con una cobertura del 99 % de la población de recién nacidos de esta comunidad. Esto representa aproximadamente 70.000 niños por año.
Todas las muestras de los hospitales públicos y privados con maternidad se envían a un único laboratorio de referencia en el Hospital Gregorio Marañón, y los casos positivos se comunican a los pediatras de la consulta de hemoglobinopatías de la misma institución. Aunque esta centralización logra una adecuada relación coste-efectividad, los controles posteriores de los enfermos se realizan en centros más cercanos al domicilio familiar.
El cribado es universal por varias razones. En primer lugar porque así es para las otras enfermedades del programa, y es rentable y de mayor eficacia para el diagnóstico precoz usar el mismo papel de filtro disponible previamente. Pero además, el coste está justificado por la opinión de varios comités expertos en la evaluación de laboratorios con una alta carga de trabajo1,13. Los cribados selectivos dirigidos sólo a población de riesgo (no universales) no resultan más efectivos en cuanto al coste si la muestra es mayor de 25.000 muestras anuales. Además, la interracialidad entre los progenitores es bastante común y no es fácil acotar un grupo de riesgo definido14,15, lo que inequívocamente llevaría a la pérdida de casos de pacientes afectados. Por último, la consideración de la raza como parámetro para la realización o no del análisis conllevaría dificultades éticas.
Un estudio piloto16 demostró que la instauración del programa era factible, y permitió crear los canales fluidos de coordinación entre el laboratorio y el grupo de clínicos necesarios para lograr una atención integrada a los niños. Se designó un responsable de la primera información telefónica a las familias, que se encargó de establecer la cita para la entrevista y la exploración del neonato. Esta primera visita comprendió una información general sobre la hemoglobina detectada, educación sanitaria inicial, anamnesis, exploración física, toma de una muestra para confirmación del diagnóstico y análisis a los padres y hermanos, consejo genético y emisión de informe. El resultado de la segunda muestra se comunicó telefónicamente, aprovechando para reafirmar la información previamente dada y remitiendo a la familia al hospital del área sanitaria de su domicilio. Los pacientes homocigotos se incluyeron en el protocolo nacional para esta enfermedad10.
Todos los miembros del grupo, incluyendo autoridades sanitarias, especialistas de laboratorio y clínicos revisaron periódicamente las bases establecidas al inicio del programa del cribado. El cuidado global del enfermo, incluyendo atención psicológica, social, y educación sanitaria integrando a enfermería especializada, se encuentran aún por desarrollar, pero el programa intentará abordarlos en un futuro. Considerando las 2 muestras de cada niño, la especificidad del análisis es del 100 %.
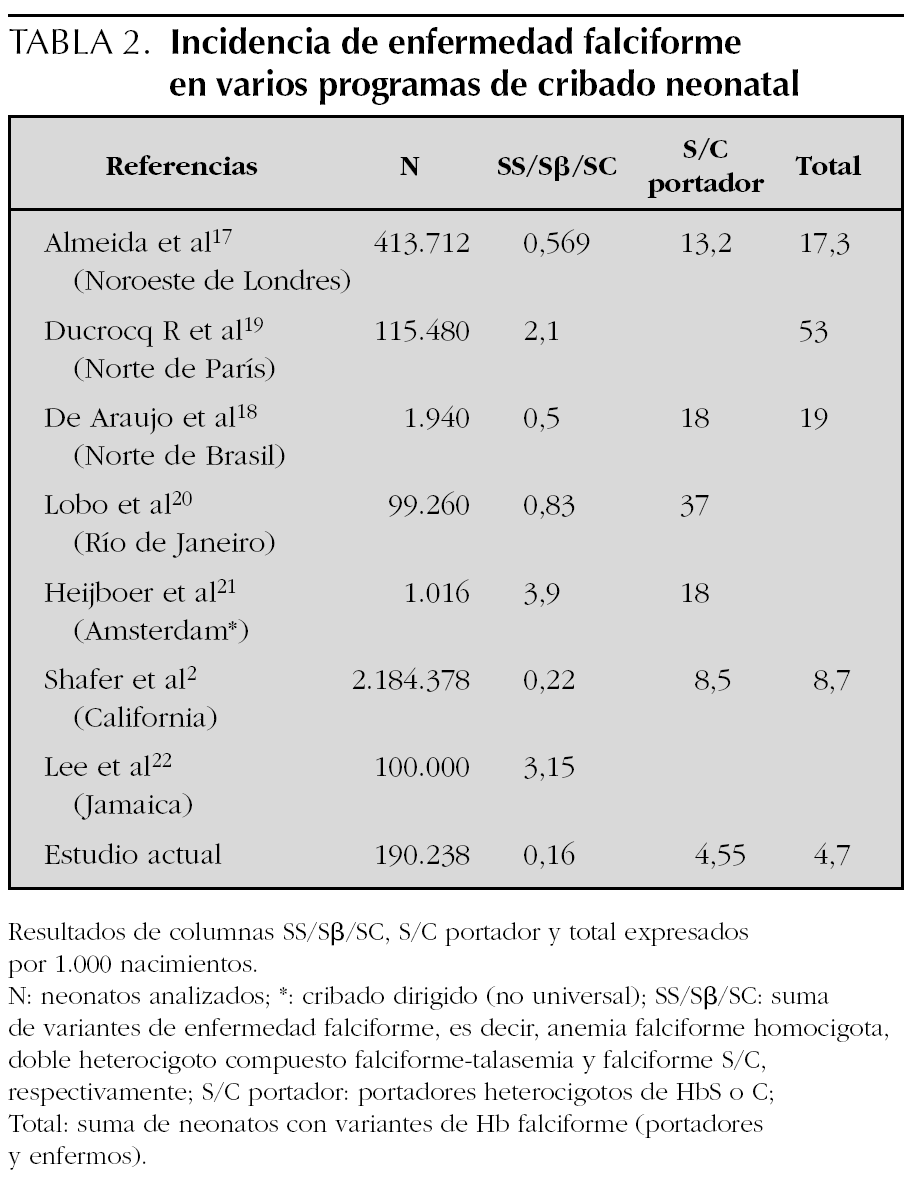
Comparando con otros programas de cribado17 (tabla 2) observamos que nuestros datos son muy similares a la prevalencia estimada en Inglaterra (0,25/1.000)1, aunque algunas regiones de ésta aumentan sus números hasta casi 0,6/1.000 nacimientos17,18 o incluso más en determinadas áreas19,21-23. Algunas fuentes2 publican una alta incidencia de variantes de hemoglobina heredadas a la vez que la HbS (como FSE, FSD, FS variante), reflejando una población diversa en cuanto a etnia. La inmigración en España ha experimentado un continuo aumento en los últimos años, y particularmente en la CM, donde 1 de cada 5 recién nacidos es de una madre extranjera (fuente: Instituto de Estadística de la CM).
Gracias al cribado neonatal se conoce la incidencia poblacional de estas patologías en la actualidad. Debido a la gran cantidad de casos detectados, se generaron problemas logísticos para la comunicación de los resultados. Considerando principalmente aspectos éticos, se decidió dar la información sobre todos los análisis alterados, incluyendo heterocigotos y variantes de hemoglobina no caracterizadas. Los beneficios para los heterocigotos son la posibilidad de recibir consejos preventivos con las recomendaciones de cuidados, consejo genético para el niño, y detección de progenitores con riesgo de tener un hijo homocigoto. Para otras alteraciones las ventajas de su conocimiento están menos definidas pero nuestra experiencia fue que la relación con la familia resultó cordial, y que en el futuro puede esperarse un beneficio añadido para su salud.
Los padres portadores son predominantemente del área caribeña y de áfrica subsahariana, pero también se han detectado árabes, indios y mediterráneos. Aunque el cribado neonatal ha sido recomendado que sea universal en áreas donde la población de minorías étnicas supere el 15 %21, es difícil estimar la proporción real de los inmigrantes con riesgo. Sólo en Madrid los inmigrantes pueden suponer cerca de 1 millón (más del 20 % de los habitantes), procedentes principalmente de América del Sur y Centro, Europa del Este y África.
La cascada de estudio familiar a raíz del caso inicial detectado hizo cuestionar la paternidad en algún caso, pero no se realizaron estudios más completos para solucionar la duda. Algunos autores24 han apuntado una tasa de no paternidad tan alta como un 10 %, pero en ausencia de preocupación de los progenitores nuestro grupo optó por considerar abiertas otras posibilidades como mutación espontánea o donación de gametos ocultada en la anamnesis.
Esperamos que este nuevo programa de cribado en Madrid disminuya la morbilidad y mortalidad asociada a la enfermedad de células falciformes, y confiamos poder probar esta hipótesis en el futuro. Además, la relación coste-efectividad debe ser igualmente examinada de forma prospectiva. La ejecución del cribado de hemoglobinopatías en otras regiones de España ha sido secundada en Extremadura, y su necesidad en el resto del territorio está por definir, aunque se están haciendo grandes esfuerzos en este sentido. La eficaz coordinación entre laboratorios, clínicos, familias y profesionales de atención primaria representa un reto enorme. El establecimiento de los consecuentes problemas éticos, necesidad de consejo antenatal y educación sanitaria pueden llevar a la sobrecarga de cualquier unidad de hematología pediátrica, por lo que es aconsejable encontrar otras vías alternativas de financiación para la colaboración con otros profesionales necesarios en el cuidado global de estos pacientes.
Agradecimientos
A todas las personas que han colaborado de forma entusiasta para que el cribado de hemoglobinopatías saliera adelante.
Investigadores del Proyecto FIS 04/0471
Además de los autores del presente trabajo, la lista de investigadores del citado proyecto que participan en tareas no comunicadas aún para esta publicación es la siguiente: Mercedes Bernácer Borja, Aurea Cervera Bravo, Rafael Díaz-Delgado y Peñas, Fernando Echávarri Olavarría, Jesús López Pérez, Celia Gil López, Elena Maderuelo Rodríguez, Arturo Muñoz Villa, Bárbara Rubio Gribble, Julián Sevilla Navarro, Jesús Saavedra Lozano, M.ª Cruz Vecilla Rivelles, Dolores Vigil Escribano.
Financiado en parte por el Fondo de Investigación Sanitaria PI 04/0471. Instituto de Salud Carlos III.
Correspondencia: Dra. E. Cela de Julián.
Departamento de Pediatría. Unidad de Hemato-Oncología.
Hospital General Universitario Gregorio Marañón.
Máiquez, 9. 28009 Madrid. España.
Correo electrónico: ecela.hgugm@salud.madrid.org
Recibido en agosto de 2006.
Aceptado para su publicación en octubre de 2006.
