


La distinción entre el hiperinsulinismo congénito (CHI) focal y difuso es esencial de cara al tratamiento y pronóstico de la enfermedad. El objetivo es presentar el primer caso de CHI focal diagnosticado en España combinando los estudios genético y PET-TC.
MétodosPaciente de 13 meses con CHI y pruebas de imagen convencionales normales, tratado con diazóxido, control dietético y alimentación por gastrostomía. Se analizó la secuencia de los genes ABCC8 y KCNJ11, y realizó una PET-TC con 18F-fluoro-L-DOPA.
ResultadosSe detectó una mutación patogénica (G111R) en el alelo paterno de ABCC8. La PET-TC demostró un foco hipercaptante en el cuerpo del páncreas compatible con un adenoma confirmado histopatológicamente. Tras la cirugía el paciente continúa asintomático sin tratamiento farmacológico ni medidas dietéticas.
ConclusionesLa combinación del análisis genético y la PET-TC con 18F-fluoro-L-DOPA muestra un gran potencial para la identificación, localización y guía de la cirugía del CHI.
Congenital hyperinsulinism (CHI) is the most common cause of persistent hypoglycaemia in infancy. The differential diagnosis between focal and diffuse forms of CHI is of great importance when planning surgery. The aim of this article is to show the first case of focal CHI diagnosed in Spain using PET-CT imaging combined with genetic analysis.
MethodsA 13 month child with CHI and normal conventional radiological investigations treated with diazoxide, diet control and feeding by gastrostomy is presented. Genetic analysis of ABCC8 and KCNJ11 genes and PET-TAC using 18F-fluoro-L-DOPA were performed.
ResultsA pathological mutation (G111R) in the paternal allele of ABCC8 was detected. PET-CT scanning using 18F-fluoro-L-DOPA showed a focus of high uptake in the body of the pancreas compatible with adenoma that was hystopathologically confirmed. After surgical resection the patient is asymptomatic without needing either pharmacological treatment or dietetic control.
ConclusionsThe combination of genetic analysis and 18F-fluoro-L-DOPA PET-TAC shows a great potential for the identification, location and guideline for surgery in CHI.
El hiperinsulinismo congénito (CHI) es la causa más común de hipoglucemia persistente en el lactante. El CHI requiere un tratamiento médico y/o quirúrgico agresivo para prevenir un daño cerebral grave e irreversible. La primera línea de tratamiento es dietética y farmacológica (diazóxido, octreótido, análogos de la somatostatina y nifepidino). Sin embargo, en los pacientes que no responden a estos tratamientos se plantea la pancreatectomía parcial o total1,2. La forma grave y neonatal generalmente resistente a la terapia farmacológica se asocia a mutaciones en los genes ABCC8 y KCNJ11. Estos genes codifican las dos subunidades del canal de potasio ATP dependiente (KATP) presente en la membrana plasmática de la célula beta. La ausencia o disfunción de estos canales conduce a la secreción continua de insulina y las hipoglucemias subsecuentes3. No obstante, se trata de una enfermedad genéticamente heterogénea que se presenta con dos formas histológicas diferentes: la forma difusa (CHI difusa) y la focal (CHI focal). Ambas formas son clínicamente indistinguibles aunque diferentes desde el punto de vista anatomopatológico y molecular. La CHI difusa constituye alrededor del 50–60 % de los casos4, presenta células beta con núcleos anormales e hipertrofiados a lo largo de todo el páncreas y suele estar asociada a la presencia de dos mutaciones, una en cada alelo del gen ABCC85. La CHI focal se presenta en el 40-50%4 y se caracteriza por la presencia de uno o varios focos de hiperplasia de células beta con núcleos anormales. Desde el punto de vista genético la forma focal se debe a un doble mecanismo, por un lado la pérdida somática durante el desarrollo fetal de la región p15 del cromosoma 11 materno, y por el otro, la presencia de una mutación en el alelo paterno del gen ABCC8 o KCNJ115.
La diferenciación entre las formas difusas y focales resulta compleja mediante técnicas de imagen convencional como la tomografía computarizada (TC) y la resonancia magnética (RM)6. Otros métodos como la estimulación arterial selectiva con calcio y la determinación de insulina por cateterismo selectivo5, son métodos invasivos y complejos con un limitado valor predictivo y, consecuentemente, una baja precisión diagnóstica7. Recientemente, se ha empleado la tomografía por emisión de positrones (PET) con L-DOPA marcada con 18Flúor (18F-fluoro-LDOPA), en pequeñas series de niños para la localización de lesiones pancreáticas causantes de CHI focal8. Los tejidos neuroendocrinos están constituidos por células capaces de captar y metabolizar precursores de aminas (células APUD). A pesar de que las células beta del páncreas no derivan del tejido neuroectodérmico mantienen la capacidad de decarboxilar la L-DOPA. Por tanto, la PET con 18F-fluoro-L-DOPA permite evaluar la decarboxilación de la L-DOPA a dopamina en estos tejidos4.
Con esta observación clínica queremos demostrar la utilidad real de la combinación del análisis genético y la PET para mejorar tanto la identificación como la localización del CHI y así optimizar el tratamiento quirúrgico.
MÉTODOSPacienteNiño de 13 meses, sin antecedentes familiares de interés, afectado de hiperinsulinismo congénito de inicio neonatal. Al nacer el peso fue de 4.510g, el test de Apgar 7/10 y no se evidenciaron malformaciones externas. En las primeras horas de vida presentó dos episodios de cianosis sin movimientos paroxísticos asociados, y determinación de glucemia capilar de 19mg/dl que requirió la perfusión intravenosa de glucosa, precisando aportes de hasta 15mg/kg/min. Tras iniciar el tratamiento con hidrocortisona (6mg/kg/día) y glucagón en momentos concentos de hipoglucemia, al sexto día de vida se introdujo el tratamiento con octreótido subcutáneo hasta una dosis de 20mg cada 6h que le causó diarrea, por lo que se sustituyó por diazóxido 30mg cada 8h, tratamiento que mantiene en la actualidad. La alimentación se realiza mediante tomas orales cada 4h y suero glucosado al 10% por gastrostomía (35ml/h) durante la noche. Los estudios de ecografía y resonancia magnética abdominal no evidenciaron alteraciones pancreáticas.
Análisis genéticoSe extrajeron muestras de sangre periférica del paciente y de sus padres con el necesario consentimiento informado. El método utilizado para el análisis de secuencia de los genes ABCC8 y KCNJ11 se describe con detalle en un artículo previo9.
Estudio PET-TCPara la realización del estudio se suspendió durante 2 días la toma de diazóxido10. A los 60min de la administración intravenosa de 66MBq de 18F-fluoro-L-DOPA, se llevó a cabo una exploración de cuerpo entero bajo sedación anestésica en un equipo PET-TC (Biograph, Siemens). El análisis de las imágenes se realizó cualitativamente y cuantificando la actividad de captación estándar máxima (SUVmáx) mediante regiones de interés sobre la cabeza, el cuerpo y la cola del páncreas, estableciéndose un índice con el foco de máxima captación8.
RESULTADOSAnálisis genéticoEl análisis de los genes ABCC8 y KCNJ11 reveló la presencia de la mutación 331G > C en heterocigosis en el exón 3 del gen ABCC8, que origina el cambio de aminoácido G111R. Dicho cambio fue detectado también en el padre del paciente.
Estudio PETEl análisis cualitativo mostró un claro depósito focal de aumento de captación de 18F-fluoro-L-DOPA en la región de cuerpo de páncreas, anatómicamente bien definido por medio del estudio TC realizado conjuntamente. Dicho foco presentó un índice captación SUVmáx de 1,5 respecto a la captación de la cola y cabeza de páncreas. Además, se apreció una captación fisiológica de 18F-fluoro-L-DOPA en los ganglios de la base del cerebro, la vesícula biliar y la eliminación renal (fig. 1).
Evolución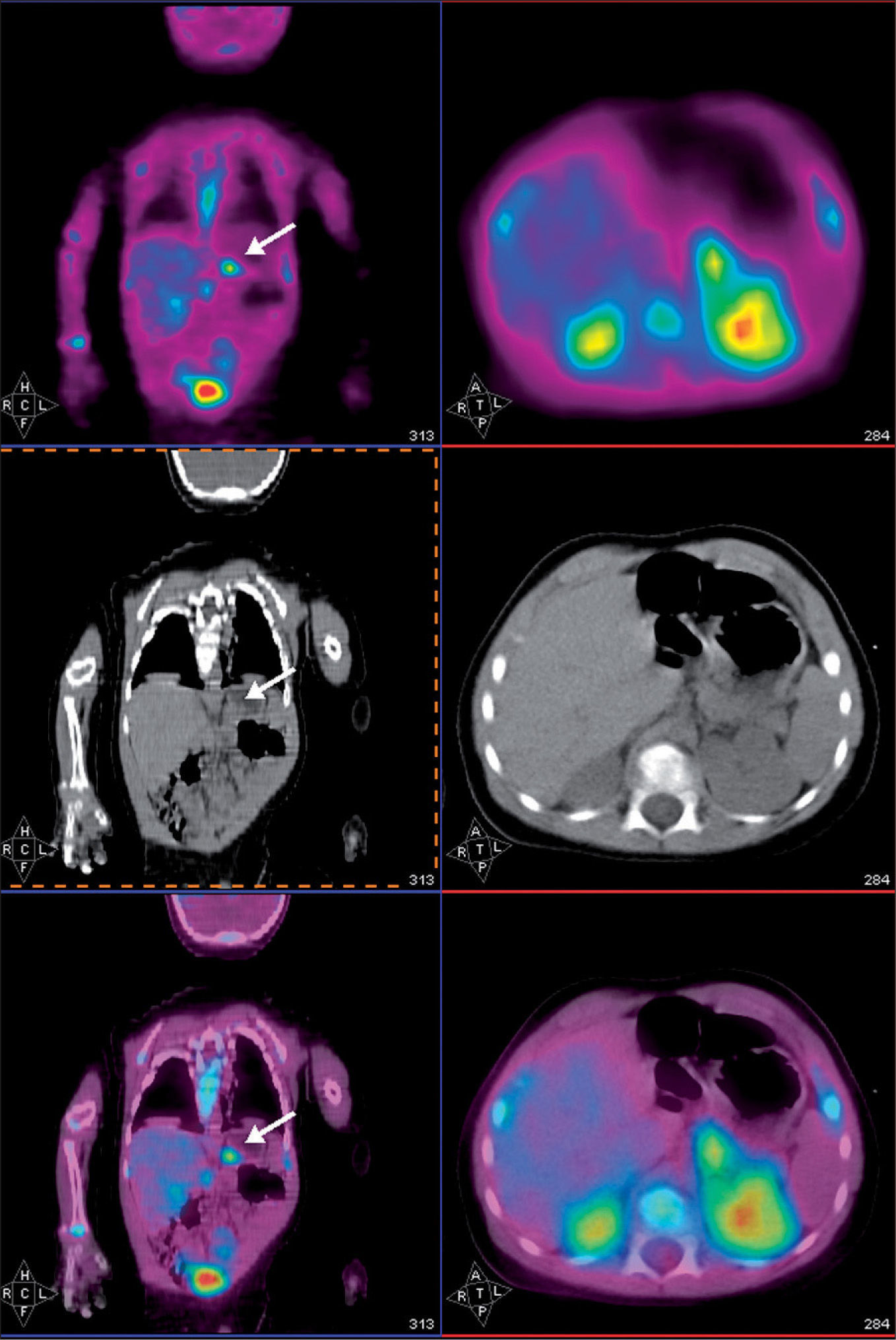
Como consecuencia de los hallazgos descritos anteriormente se procedió a la exéresis quirúrgica de la lesión apreciándose en el estudio histopatológico la existencia de un adenoma en la región de cuerpo coincidente con la localización descrita en el estudio PET-TC. Tras 5 meses de evolución, el niño se encuentra asintomático, sin necesidad de tratamiento farmacológico ni medidas dietéticas.
DISCUSIÓNLa diferenciación entre las formas de CHI focal y difusa es esencial, ya que la estrategia terapéutica, el pronóstico y, en su caso, el posible consejo genético difieren radicalmente5,11. La intervención quirúrgica puede ser necesaria para el control de la hipoglucemia pero sólo es curativa en los pacientes con CHI focal si se realiza una resección completa de la lesión pancreática. Todo ello acrecienta la necesidad de una precisa localización preoperatoria. El análisis genético del paciente presentado aquí detectó una única mutación G111R en el alelo paterno de ABCC8, la cual ya había sido descrita previamente en nuestra población8 y su patogenicidad previamente demostrada12. El estudio genético, por tanto, encajaba con un CHI focal. En el CHI la ausencia de detección de una mutación en el alelo materno, a priori, no permite descartar que ésta realmente exista, ya que la sensibilidad del método utilizado no es del 100 %. En estas circunstancias no podemos asegurar la focalidad de la enfermedad basándonos únicamente en el análisis genético. Por tanto, la realización del estudio PET con 18F-fluoro-L-DOPA resultó definitiva tanto para confirmar la focalidad como para localizar el adenoma de páncreas causante de su CHI y plantear un abordaje quirúrgico curativo.
Los focos de hiperinsulinismo infantil contienen islotes de células hiperfuncionantes muy diferenciados, mientras que los insulinomas de adultos contienen células relativamente muy poco diferenciadas. Asimismo, los islotes normales adyacentes a la lesión CHI focal se encuentran en estado basal, algo que no ocurre en los adyacentes a un insulinoma13. Por tanto, la identificación de focos hiperfuncionantes mediante una característica metabólica específica, como es la captación de L-DOPA, resulta más factible en la CHI focal de la infancia que en insulinomas de adultos. En este sentido, tiene interés realizar una estimación cuantitativa de la actividad del foco de hipercaptación de 18F-fluoro-L-DOPA respecto al resto de áreas del páncreas.
Los hallazgos presentados aquí coinciden con las recientes publicaciones en las que se demuestra la utilidad del estudio PET con 18F-fluoro-L-DOPA en la detección de la hiperplasia focal de células beta del páncreas en la CHI8,14. No obstante, la realización del estudio en un equipo híbrido PET-TC aporta una precisa correlación anatomofuncional que, en este caso, facilitó la resección quirúrgica de un adenoma confirmado histopatológicamente.
En un análisis genético previo realizado en nuestro país quedó demostrado que en al menos el 70 % de los casos de CHI se detecta mutación o mutaciones en ABCC89. En el 40 % de estos casos se detectan dos mutaciones, una en cada alelo de ABCC8, lo que determina el tipo histopatológico de la enfermedad CHI difuso. En el 30 % restante se detecta una única mutación, el 75 % en el alelo paterno y el 17 % en el materno. De este modo podemos asegurar una CHI difusa tanto en los pacientes con dos mutaciones como en los pacientes con mutación materna y acotar los posibles casos focales a los pacientes con una única mutación paterna. Precisamente en este último grupo y en aquéllos en los que no se hayan detectado mutaciones, el estudio PET y, sobre todo, PET-TC con 18F-fluoro-L-DOPA puede ofrecer el máximo rendimiento en la identificación y localización de la CHI focal. Los excelentes resultados demostrados por la PET con 18F-fluoro-L-DOPA hacen sospechar que puede ser la técnica de referencia para la guía de la resección quirúrgica en la mayoría de los casos de CHI. No obstante, el potencial diagnóstico final de esta técnica debería ser confirmado tras analizar series de pacientes más numerosas estudiadas en múltiples centros.
Los autores agradecen al doctor Peñuelas y la doctora Quincoces junto con el personal del laboratorio PET su apoyo técnico, y al personal del centro PET del Servicio de Medicina Nuclear, de la UCI Pediátrica y Servicio de Anestesia de la CUN por su excelente ayuda en la ejecución del estudio de imagen PET-TC. También a los doctores Gussinye y Chamorro de los departamentos de Pediatría del Hospital Vall d'Hebron y Complejo Hospitalario Universitario de Vigo, respectivamente, por el control y seguimiento del paciente.
