






La esclerosis tuberosa (ET) es una enfermedad de afectación multisistémica y gran variabilidad fenotípica. Están identificados 2 genes involved en la génesis de la enfermedad: TSC1 y TSC2.
ObjetivosCaracterizar clínicamente a los pacientes con ET seguidos en Neurología Pediátrica de un hospital de tercer nivel durante los últimos 10 años y correlacionar el genotipo con la gravedad de la clínica neurológica y los estudios de imagen.
Pacientes y métodosEstudio retrospectivo descriptivo, mediante consulta de la historia clínica y evaluación de las resonancias magnéticas (RM) de pacientes con ET.
ResultadosSe estudiaron 35 casos, con una mediana de edad, al diagnóstico, de 10 meses. En el 91,4% se registraron crisis epilépticas, con un predominio de espasmos epilépticos a la presentación.
Más del 50% tenía deterioro cognitivo y el 49% trastornos de conducta.
Se sometieron a estudio genético 24 niños con predominio de mutaciones TSC2 (58,3%). De los 11 casos de epilepsia refractaria, 6 tenían mutación del gen TSC2. De los 8 pacientes con déficit cognitivo moderado a grave, se identificaron 5 mutaciones TSC2.
Se revisaron 26 RM y en el 76,9% se observó una afectación completa de los lóbulos cerebrales, lo que refleja una gran cantidad de lesiones. De los enfermos con mutaciones TSC2 y RM realizada, todos tenían alta carga de lesión y 5 epilepsia refractaria.
DiscusiónEn nuestra muestra, nos encontramos con un alto porcentaje de mutaciones en el gen TSC2. Esta mutación está asociada a un peor pronóstico neurológico, con crisis más farmacorresistentes y un atraso cognitivo más severo.
Tuberous sclerosis (TS) is an inherited disorder with multisystemic involvement and a high phenotypic variability. There are two genes that cause this condition: TSC1 and TSC2.
ObjectivesOur goal was to clinically characterize patients with TS followed up in the Pediatric Neurology Clinic of a tertiary hospital during the last 10 years, and correlate the genotype with the severity of neurological manifestations and imaging studies.
Patients and methodsRetrospective analysis of patients with TS, including review of medical records and available MRI imaging.
ResultsWe studied 35 cases with a median age at diagnosis of ten months. Seizures were the first manifestation in 91.4% of cases, with a predominance of epileptic spasms. Over 50% had cognitive impairment and 49% behavioral disorders.
A genetic study was performed on 24 children, and TSC2 mutations identified in 58.3% of them. Of the 11 cases of refractory epilepsy, six had the TSC2 gene mutation. In the group of eight patients with moderate/severe cognitive deficits, five had TSC2 mutations.
We reviewed 26 MRI scans, in which it was observed that 76.9% had diffuse involvement of cerebral lobes, which reflects a greater burden of injury. Of the patients who had an MRI scan performed and had TSC2 mutations, all had a high tuber load, and5 of them had refractory epilepsy.
DiscussionIn our sample we observe a high percentage of mutations in the TSC2 gene. This mutation carries a worse neurological prognosis, with drug-resistant epilepsy and a more severe cognitive impairment.
La esclerosis tuberosa (ET) es un síndrome neurocutáneo multisistémico. Las alteraciones observadas resultan de una disfunción de la diferenciación, proliferación y migración celulares en las primeras etapas del desarrollo fetal1.
La ET tiene una herencia autosómica dominante, con una incidencia de aproximadamente uno por cada 5.000 a 10.000 nacimientos2. Actualmente, se han identificado mutaciones en 2 genes implicados en la génesis de la enfermedad: TSC1 (cromosoma 9q34) y TSC2 (cromosoma 16p13.3). Solo el 7-37% tienen antecedentes familiares positivos, siendo que la mayoría (65-75%) corresponden a mutaciones «de novo»3. Presentan una gran variabilidad de expresión fenotípica, edad de aparición, signos, síntomas y gravedad.
En el 60-89% de los enfermos que cumplen los criterios de diagnóstico para ET se identifica una mutación causante de la enfermedad, siendo que se estima que alrededor del 50% corresponden a mutaciones en TSC2 y el 17% a mutaciones en TSC14–6.
La ET se caracteriza por la aparición de tumores benignos en múltiples órganos7. Además, hay un aumento del riesgo de malignidad8.
El diagnóstico se define únicamente por criterios clínicos (tabla I). Se clasifica como ET definitiva si están presentes 2 criterios mayores o uno mayor y 2 menores; ET probable si se registran uno mayor y uno menor, y ET posible cuando se observa uno mayor o 2 o más menores, sin criterios mayores. La evaluación genética es útil para estudios de la familia o de confirmación de posibles o probables casos de ET.
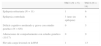
Criterios de diagnóstico de esclerosis tuberosa: Roach
| Criterios mayores | Criterios menores |
| Angiofibromas faciales/placa fibrosa | Lesiones cutáneas en «confetti» |
| Placas de Shagreen | Fibromas gingivales |
| ≥ 3 manchas hipopigmentadas | Lesiones del esmalte dentario |
| Fibromas periungueales o ungueales | Pólipos rectales hamartomatosos |
| Linfangiomiomatosis | Quistes renales múltiples |
| Angiomiolipoma renal | Hamartomas no renales |
| Rabdomioma cardiaco | Quistes óseos |
| Hamartomas nodulares múltiples retinianos | Hamartoma retiniano |
| Tuberomas corticales | Líneas de migración radiales de la sustancia blanca |
| Nódulos subependimarios | |
| Astrocitomas de células gigantes subependimarios |
Los síntomas neurológicos, presentes en el 85% de los casos, son la principal causa de morbimortalidad3. La epilepsia y el retraso cognitivo están comúnmente asociados a lesiones cerebrales, incluyendo hamartomas glioneuronales (también llamados tuberomas), lesiones de la sustancia blanca y astrocitomas de células gigantes subependimarios9.
La epilepsia aparece generalmente en el primer año de vida y una forma de presentación frecuente son los espasmos epilépticos (36-96%)10. El número y, en particular, el volumen total ocupado por hamartomas glioneuronales están relacionados con la presencia de disfunción cerebral grave (epilepsia refractaria y/o deterioro cognitivo moderado a grave)1,11. También se ha demostrado la asociación entre el grado de gravedad de la epilepsia y la presencia de tuberomas corticales con aspecto quístico12.
Se estima que el 50% de los enfermos tienen afectación cognitiva y su gravedad se asocia a los antecedentes de espasmos epilépticos o epilepsia refractaria y al número y el volumen ocupado por los hamartomas glioneuronales13.
Los problemas de conducta están presentes en un 40-90% de los pacientes con ET. Aunque los cambios comportamentales puedan ocurrir independientemente del nivel cognitivo, se verifica que la discapacidad intelectual y la elevada frecuencia de crisis epilépticas son factores de riesgo para la existencia de trastornos de conducta14.
Las manifestaciones dermatológicas pueden ocurrir en el 81-95% de los casos de ET, siendo las más comunes: manchas hipopigmentadas, angiofibromas, fibromas ungueales, placas de Shagreen, placas fibrosas10,15. No existe un riesgo significativo de transformación maligna de lesiones cutáneas.
Entre el 50-60% de los enfermos con ET tienen afectación cardiaca, siendo el rabdomioma la más común. Se trata de un tumor benigno, a menudo multifocal, normalmente asintomático y en general con regresión espontánea.
La afectación renal, presente en el 60-80% de los enfermos con ET, tiene como forma más común de presentación el angiomiolipoma. Las lesiones generalmente son benignas, con un riesgo de hemorragia asociado al tamaño de la lesión. Presentan, además, un alto riesgo para desarrollar hipertensión dependiente de renina y enfermedad renal crónica, debido a la compresión y la sustitución del parénquima renal. El carcinoma de células renales surge en aproximadamente el 1-2% de los adultos, por lo que se recomienda un control regular por ecografía.
La afectación oftalmológica está presente en el 87% de los enfermos con ET. A pesar de útiles en el diagnóstico, rara vez afectan a la agudeza visual y no requieren tratamiento específico16.
Con la identificación de los 2 genes asociados a ET, se viene estableciendo una correlación entre el genotipo y el fenotipo de la enfermedad. Varios estudios consideran que mutaciones del gen TSC1 están asociadas a un fenotipo menos grave que las del TSC2, pero esta relación no es consensual15-17. Aunque las mutaciones en TSC2 tienden a asociarse con fenotipos más graves (con una mayor incidencia de epilepsia refractaria, deterioro cognitivo y cambios de comportamiento), también se han descrito fenotipos leves de mutación en TSC217.
ObjetivosCaracterizar clínicamente a los enfermos con ET seguidos en consulta de Neurología Pediátrica del Centro Hospitalario del Porto (CHP) en los últimos 10 años y correlacionar el genotipo con la severidad de las manifestaciones neurológicas y de los hallazgos de imagen.
Pacientes y métodosAnálisis descriptivo retrospectivo de los enfermos con diagnóstico definitivo de ET (de acuerdo con los criterios clínicos de la enfermedad), seguidos en consulta de especialidad de Neurología Pediátrica en un centro terciario, el CHP, Porto, Portugal. Los datos se obtuvieron mediante la consulta de los registros electrónicos de salud y de los procesos clínicos en papel.
El programa estadístico utilizado ha sido el Microsoft Office Excel 2007 para las estadísticas descriptivas. Las variables analizadas han sido la edad, el sexo, la edad en el momento del diagnóstico, la historia familiar, la epilepsia y su caracterización, el desarrollo psicomotor, los cambios de comportamiento, cutáneos, cardiacos, renales y oftalmológicos, y el estudio genético.
Las resonancias magnéticas (RM) cerebrales evaluadas han sido revisadas de forma independiente por el mismo examinador de la especialidad de Neurorradiología y se estudiaron las siguientes variables: número de tuberomas, localización supra o infratentorial, el número de lóbulos cerebrales con lesiones, el número y la lateralización de tuberomas con quistes, las líneas de migración radial de la sustancia blanca, los nódulos subependimarios y los astrocitomas de células gigantes. Los pacientes efectuaron los estudios de RM en equipos GE Signa 1.5T y en RM Philips Achieva 3.0T TX (3 pacientes), en el Departamento de Neurorradiología del CHP. Después de obtenida la imagen localizadora en el plano sagital, para identificar las comisuras anterior y posterior, se han adquirido imágenes en paralelo al plano intercomisural: FLAIR, T2 SE, T2 *, T1 SE y T1 SE tras contraste.
Para la identificación de tuberomas y líneas de migración radial han sido utilizadas imágenes FLAIR18 y para la identificación de los nódulos subependimarios, imágenes FLAIR y ponderadas en T2.
Se definió tuberoma como una lesión hiperintensa corticosubcortical en FLAIR y líneas de migración radiales como hiperintensidades lineales en FLAIR, que se extiende desde la superficie ventricular hasta el tuberoma cortical. Los nódulos subependimarios han sido investigados en cualquier ventrículo, con origen en la pared ventricular y con proyección hacia el interior de los mismos.
Se consideraron tuberomas con quistes subcorticales las lesiones con una señal similar al del líquido cefalorraquídeo en todas las ponderaciones y con un anillo hiperintenso alrededor en T2. Los tuberomas o nódulos subependimarios calcificados han presentado una hiposeñal en T2 y T2*. Una vez que la cantidad o el número de tuberomas en el parénquima encefálico son indicados como el mejor marcador de gravedad de epilepsia y de la función cognitiva1, se efectuó una evaluación cualitativa por medio la frecuencia de tuberomas. En cada lóbulo cerebral, la densidad de tuberomas se relaciona con la carga de lesión cerebral obtenida por la evaluación del número total de tuberomas y de la afectación de cada lóbulo por los tuberomas. Se consideró como elevada carga de lesión la afectación de todos los lóbulos por tuberomas, en pacientes con número total de tuberomas más de 15. No han sido evaluados la densidad o el volumen de tuberomas por técnica cuantitativa de segmentación automática de tuberomas por inaccesibilidad a software apropiado.
La evaluación cognitiva formal se realizó según el grupo de edad a través de la Escala de desarrollo mental de Ruth Griffiths y/o Escala de Wechsler —WISC-R o WISC-III en la consulta de Psicología del CHP. El déficit cognitivo se definió como coeficiente intelectual (CI)<70, con los siguientes niveles de gravedad: ligero, con un CI entre 50 a 55-70; moderado 35 a 40-50 a 55; severo 20 a 25-35 a 40, y profundo <20 a 25. Se clasificó como déficit cognitivo «no especificado» cuando este no ha sido examinado pero que se presume bajo (o sea, <70)19.
Los exámenes genéticos se realizaron en 2 laboratorios especializados: en el Centro de Genética Médica, Porto, a través del Programa de Erasmus MC University Medical Center Rotterdam, por análisis de secuenciación directa y del test Multiplex Ligation-dependent Probe Analysis y en el Centro de Genética Clínica, Porto, por PCR y secuenciación directa de toda la región codificante, incluso las regiones intrónicas adyacentes, de los genes TSC1 y TSC2; se realizó, además, PCR cuantitativa para estudio de deleciones en el gen TSC2.
Epilepsia refractaria se definió como epilepsia no controlada con 2 o más fármacos antiepilépticos en dosis apropiada y adecuada al tipo de crisis20.
ResultadosSe analizaron 35 casos con diagnóstico definitivo de ET, caracterizados en la tabla II.
Características clínicas de 35 pacientes con esclerosis tuberosa
| % (N) | Información adicional | |
| Género masculino/femenino | 60,0 (21)/40,0 (14) | |
| Grupo de edad actual mín/máx/mediana | 8 meses/28 años/12 años | |
| Edad de diagnóstico mín/máx/mediana | Prenatal/7 años/15 meses | |
| Diagnóstico en 1.er año de vida | 65,7 (23) | 82,9% (29) en los 2 primeros años |
| Estudio genético positivo | 79,2 (19)/24 | |
| TSC2/TSC1 | 58,3 (14)/20,8 (5) | |
| Historia familiar positiva | 25,7 (9) | 36,8% (7) en el grupo de mutación |
| Principal motivo de referencia-epilepsia | 77,1 (27) | 20,0% (7) rastreo neurológico ante la sospecha de ET |
| Epilepsia | 91,4 (32) | Espasmos epilépticos, 53,1% (17)Crisis focales, 34,4% (11)Crisis generalizadas, 6,3% (2)71,9% (23) crisis en el 1.er año de vida34,4% (11) epilepsia refractaria |
| Nivel cognitivo | 57,1 (20) | Ligero 50,0% (10)Moderado 25,0% (5)Grave 15,0% (3)No especificado pero aparentemente con CI<70-10,0% (2) |
| Trastornos de conducta | 48,6 (17) | Conducta oposicionista 5Hiperactividad e/o inatención 5Agresividad 4Estereotipias y/o manerismos 3Trastorno del espectro autismo 1 |
| Cardiaco | 42,9 (15) | Rabdomiomas 93,3% (14)Multifocales 8/14Arritmia 2 (uno con rabdomioma multifocal) |
| Renal | 42,9 (15) | Angiomiolipomas 80,0% (12)Quistes benignos 53,3% (8), 5 asociados a angiomiolipomasHTA 1 |
| Oftalmológico | 20,0 (7) | Hamartomas retinianos 4Lesiones no retinianas 2Despigmentación coriorretiniana 1 |
| Dermatológico | 74,3 (26) | Manchas hipopigmentadas 80,8% (21), 11 asociados a angiofibroma y uno con placas fibrosasAngiofibroma aisladamente 5 |
Veinticuatro niños realizaron estudio genético y se identificó mutación en el 79,2% (19) de los casos, siendo la mutación en el gen TSC2 la más frecuente (58,3%), en relación con la del gen TSC1 (20,8%).
La historia familiar ha sido positiva en 25,7% (9): 3 con un único familiar de primer grado afectado, 2 con 2 familiares afectados y 4 con el diagnóstico a posteriori en familiares de primer grado (padres o hermanos).
En el grupo en que ha sido identificada la mutación, el 36,8% presentaba historia familiar positiva; en los casos con mutación en TSC1, el 40,0% (2/5) y en los casos con mutación en TSC2, el 35,7% (5/14).
Los motivos de derivación para la consulta de Neurología Pediátrica fueron: epilepsia en el 77,1% (27) de los casos, bien como uno por retraso en el desarrollo psicomotor asociado a alteraciones en la RM y 7 para rastreo de afectación neurológica en niños con sospecha de ET (6 casos de hamartomas cardiacos, 2 de los cuales asociados a alteraciones cutáneas y uno con historia familiar y alteraciones cutáneas).
En el 91,4% (32) de los enfermos han sido registradas crisis epilépticas. Como forma de presentación, predominaron los espasmos epilépticos en el 53,1% (17). En el 71,9% (23) de los casos las crisis epilépticas han empezado en el primer año de vida, con un 34,4% (11) de enfermos con epilepsia refractaria.
En los enfermos con epilepsia controlada, actualmente 11 toman un fármaco antiepiléptico, 2 toman 2 fármacos y 7 no siguen medicación. Solamente un caso ha sido sometido a cirugía, con una buena respuesta. En el tratamiento de los espasmos epilépticos, la vigabatrina ha sido el fármaco de elección (88,2%), en monoterapia o en asociación.
El déficit cognitivo ha sido documentado en el 57,1% (20) de los niños y los trastornos de conducta descritas en 48,6% (17).
El 42,9% (15) presentó afectación cardiaca, el 42,9% (15) manifestaciones renales y el 20,0% (7) alteraciones oftalmológicas.
Las manifestaciones dermatológicas se identificaron en el 74,3% (26) y no se observó ningún caso de transformación maligna.
Correlacionando genotipo y clínica (tabla III), se asociaron 6 de los 11 casos de epilepsia refractaria a la mutación en TSC2 y solo un caso a la mutación en TSC1.
Correlación genotipo/fenotipo
| TSC1 (N=5) | TSC2 (N=14) | |
| Epilepsia refractaria (N=11) | 1 | 6 |
| Epilepsia controlada | 3 (uno sin epilepsia) | 8 |
| Déficit cognitivo moderado y grave con estudio genético (N=6/8) | 0 | 5 |
| Alteraciones de comportamiento con estudio genético (11/17) | 0 | 9 |
| Elevada carga lesional en la RM | 1 | 13 |
En los restantes casos con mutación en TSC1, hubo un caso sin epilepsia y 3 con epilepsia controlada. En los otros casos con mutación en TSC2, se encontró a 8 pacientes con epilepsia controlada (6 con un fármaco y 2 sin fármacos).
De los casos 8 con déficit cognitivo moderado y grave (8), 6 realizaron estudio genético, siendo identificadas mutaciones en el gen TSC2 en 5 enfermos, sin encontrar casos asociados a mutación en TSC1. Además, de los 11 casos con trastornos del conducta que realizaron estudio genético, 9 presentaron mutación en TSC2, no existiendo ningún caso con mutación en TSC1.
De los 8 casos de retraso en el desarrollo cognitivo moderado y grave, 5 presentaron epilepsia con inicio antes del 12 meses de vida. Los 3 casos de retraso grave presentaban epilepsia refractaria y perturbaciones de comportamiento. Sin embargo, 3 de los 16 casos con epilepsia de inicio precoz y espasmos epilépticos presentaron un desarrollo psicomotor normal (evaluación cognitiva formal realizada a los 3, 7 y 11 años de edad).
El 74,3% (26) de las RM disponibles han sido revisadas en todos los lóbulos cerebrales en el 76,9% (20) de los pacientes, en los cuales se identificaron más de 15 tuberomas, es decir, una elevada carga de lesión. En el 96,2% (25) de los enfermos se observaron líneas de migración radial (fig. 1). Estaban presentes nódulos subependimarios en el 84,6% (22), la mayoría de los cuales con calcificación y, en 3 casos, con captación de contraste (fig. 2). En el 23,1% (6) de los enfermos se encontraron tuberomas infratentoriales. Siete casos presentaban tuberomas con aspecto quístico (26,9%), 6 de los cuales en localización yuxtacortical y uno infratentorial (fig. 3). Es de referir un caso con probable astrocitoma de células gigantes, bajo control dimensional, en la región del agujero de Monro, sin signos de hidrocefalia (fig. 4). Ninguno de los enfermos presentaba atrofia focal.
 y líneas de migración radial.")



De los enfermos con mutación en TCS2 identificada, solamente en uno la RM no ha sido revisada, presentando todos los restantes una elevada carga de lesión. Uno de 5 pacientes con mutación TSC1 presentó elevada carga de lesión. De los 5 casos asociados a epilepsia refractaria, 2 presentaron tuberomas con alteraciones quísticas.
El 87,5% de los enfermos con retraso cognitivo y el 78% con epilepsia de inicio anterior a los 12 meses presentaron elevada carga de lesión. En este grupo con epilepsia de inicio precoz, el 89% ha revelado, también, líneas de migración radial.
DiscusiónLa ET es una enfermedad multisistémica con expresión fenotípica variable y manifestaciones clínicas a veces sutiles, poco específicas y de aparición en diferentes edades, por lo que frecuentemente se retrasa el diagnóstico21. En esta serie, el diagnóstico ha sido precoz, probablemente porque el principal motivo de referencia para la consulta era espasmos epilépticos, que han sido el principal motivo de referencia para la consulta; esto ha facilitado su diagnóstico, de forma asociada a la sensibilización para esta afección, por parte de los profesionales de salud.
En el grupo de niños que han realizado estudio genético, se identificaron mutaciones en el 79,2%, con mayor prevalencia de mutaciones en el gen TSC2, dato que está de acuerdo con otros estudios4-6.
El número de casos con historia familiar positiva es bajo3, pero superior en el grupo de enfermos con mutación identificada. En 4 casos, la historia familiar solo se identificó posteriormente al diagnóstico en familiares de primer grado (padres o hermanos), lo que refuerza la importancia de evaluar los criterios clínicos de ET en los padres o hermanos de niños con diagnóstico de ET sin historia familiar positiva22.
Como ya ha sido descrito, la epilepsia ha sido el principal motivo de referencia1, manifestándose, en la mayoría de los casos, en el primer año de vida y con espasmos epilépticos.
Se correlacionó positivamente la gravedad y la refractariedad de la epilepsia con la edad precoz del inicio de las crisis: todos los casos de epilepsia refractaria iniciaron crisis antes de los 12 meses de edad.
La vigabratina ha sido el fármaco más utilizado (88,2%) en el tratamiento de los espasmos en nuestros enfermos con ET, lo que está de acuerdo con el hecho de ser la opción más eficaz en esta situación23, referida por investigadores del National Institute of Health Tuberous Sclerosis Complex Consensus Conference (2000).
Ha sido documentada la presencia de déficit cognitivo en un porcentaje elevado de casos, con gravedad muy variable, dato en conformidad con lo descrito (44-65%) y que demuestra la importancia de la realización de una evaluación cognitiva formal inicial en todos los casos de ET, para la implementación de programas de intervención precoz adecuados en niños afectados, de forma a programar la intervención y estimulación cognitivas, buscando mejorar el pronóstico.
Nuestros resultados confirman la existencia de una correlación positiva entre el retraso en el desarrollo psicomotor (RDPM) y la epilepsia, presentándose una epilepsia con inicio en el primer año y evolución para epilepsia refractaria en todos casos de retraso cognitivo grave9. Por otro lado, en nuestra muestra se han encontrado casos de espasmos epilépticos precoces y epilepsia refractaria sin RDPM. Los trastornos de conducta han sido muy frecuentes y se asociaron a retraso cognitivo y epilepsia refractaria.
En nuestra serie, se confirmó que la principal característica cardiaca de ET es el rabdomioma (14/15), con un predominio multifocal (8/14). El porcentaje de envolvimiento renal y oftalmológico ha sido inferior al descrito, y sin registro de complicaciones significativas (solo un caso de hipertensión arterial), probablemente por tratarse de una muestra en edad pediátrica. Se registró envolvimiento dermatológico en un porcentaje inferior al descrito habitualmente, hecho que relacionamos con la pobre descripción de estas alteraciones en los registros clínicos.
Se verificó un peor pronóstico neurológico de las mutaciones en TSC2, con mayor porcentaje de epilepsia farmacorresistente, retraso cognitivo más grave, mayor frecuencia de perturbaciones de comportamiento y mayor carga de lesión en la RM.
El 87,5% de los enfermos con retraso cognitivo y el 78% con epilepsia en edad temprana presentaron una elevada carga lesional, corroborando la asociación demostrada en varios estudios entre la epilepsia, la función cognitiva y la carga lesional1.
En el grupo de enfermos estudiados con RM, las líneas de migración radial han sido la alteración radiológica más frecuente (96,2%), lo que está próximo del resultado de 100% descrito por Van Eeghen et al. en literatura reciente24. Este grupo describe, además, una asociación entre las líneas de migración radial y la precocidad de la edad de inicio de las crisis epilépticas, hecho también confirmado en nuestra muestra.
Esta serie es de una muestra pediátrica, sensibilizando para las principales comorbilidades y manifestaciones de la enfermedad, para la evolución y nivel de gravedad de los enfermos con ET en este grupo de edad, y ayudando a optimizar la práctica clínica.
Como limitaciones del trabajo, se realza el hecho de tratarse de un estudio retrospectivo, dependiente de registros clínicos, y de ser realizado en un centro terciario, seleccionando los casos más graves; aun más, es de destacar la falta de uniformidad en relación con la evaluación clínica inicial y el seguimiento entre los diferentes especialistas, bien como el tamaño y la heterogeneidad de la muestras, que interfiere en la significación estadística de las conclusiones.
La realización de estudios multicéntricos en el futuro permitirá uniformizar su seguimiento.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
