

El déficit selectivo de IgA (DSIgA) es la inmunodeficiencia primaria más frecuente, siendo a menudo asintomática. Se ha descrito una elevada agregación familiar, sin conocerse el defecto genético causante ni su mecanismo hereditario.
ObjetivosDefinir la utilidad del cribado de los familiares de primer grado de los pacientes con DSIgA valorando si los casos familiares presentan unas características clínicas e inmunológicas más graves que los casos esporádicos (CE) y si los familiares diagnosticados de DSIgA presentan sintomatología clínica significativa para justificar su cribado.
Pacientes y métodosEstudio transversal descriptivo (octubre del 2010-septiembre del 2011) de todos los pacientes con DSIgA controlados en nuestro centro, con revisión de datos demográficos, clínicos y analíticos. Se consideró como caso familiar (CF) todo aquel con al menos un familiar de primer grado (FPG) con DSIgA.
ResultadosDe los 130 participantes, 42 eran pacientes con DSIgA y 88 FPG. Se diagnosticaron 13 CF (31%), 29 CE (69%) y 14 (16%) FPG enfermos (FPG-E). El número necesario a analizar para encontrar un FPG-E fue de 6 familiares. No hubo diferencias clínicas entre los pacientes. Hubo una proporción mayor de patología intestinal (p=0,001, OR=9,57, IC del 95%, 2,59-35,3), ingresos (p=0,045, OR=4,01; IC del 95%, 1,10-14,67) y necesidad de tratamiento crónico (p=0,006, OR=5,5; IC del 95%, 1,57-19,54) en los FPG-E con respecto a los FPG sanos.
ConclusionesA pesar de no encontrar más complicaciones clínicas en los CF de DSIgA, la elevada prevalencia de familiares afectados con afectación clínica significativa podría justificar la realización sistemática de estos programas de cribado.
Selective immunoglobulin A deficiency (SIgAD), the most common primary immunodeficiency, is often asymptomatic. High rates of familial clustering have been described in SIgAD, but the causative genetic defect and mechanism of inheritance are unknown.
ObjectivesTo determine whether familial SIgAD cases show more severe clinical and immunological characteristics than sporadic ones; to investigate the utility of screening first-degree relatives (FDRs) of these patients, and to determine whether symptoms in affected family members are important enough to justify screening.
Patients and methodsDescriptive, cross-sectional study (October 2010-September 2011) of all patients with SIgAD and followed up in our center. Demographic, clinical, and analytical data were reviewed. A familial case was defined as an SIgAD patient with at least one affected FDR.
ResultsOf the 130 participants, 42 were SIgAD patients and 88 FDR. There were 13 (31%) familial cases and and 14 (16%) affected FDRs. Six family members had to be analyzed in order to detect one affected one. There were no clinical differences between familial and sporadic SIgAD cases. The percentages of intestinal disease (p=001, OR=9.57, 95%CI 2.59-35.3), hospitalizations (p=045, OR=4.01; 95%CI 1.10-14.67], and need for chronic treatment (p=006, OR=5.5; 95%CI 1.57-19.54) were higher in affected FDRs than in unaffected ones.
ConclusionsThe symptoms were not more severe in familial than sporadic SIgAD cases. Nonetheless, the elevated prevalence of affected FDRs with significant morbidity may justify routine screening of close family members of these patients.
El déficit selectivo de IgA (DSIgA) pertenece al grupo de inmunodeficiencias primarias caracterizado por un déficit en la producción de anticuerpos. Aunque el DSIgA es la inmunodeficiencia primaria (IDP) más frecuente, su prevalencia puede variar desde 1/163 a 1/18.500 casos en función de la población estudiada y de la definición aplicada como DSIgA1,2. Estas variaciones apoyan una predisposición genética, pudiendo aparecer nuevos casos de forma esporádica o ser transmitido de forma autosómica dominante, recesiva o mediante herencia poligénica. Así, se ha estimado un riesgo del 30% de probabilidad de padecer DSIgA si hay un familiar afectado3, pero no se ha identificado un defecto genético concreto ni se ha comprobado un patrón de herencia mendeliano claro, manteniendo una expresividad y penetrancia variables2,4,5. Se han identificado varios genes posiblemente implicados en su etiopatogenia, lo que hace pensar que el DSIgA es probablemente un grupo heterogéneo de enfermedades y/o de anomalías genéticas, de una manera parecida a la inmunodeficiencia común variable (IDCV) entre los que destacan transmembrane activator and calcium modulator (TACI), B-cell activating factor (BAFF-receptor), proliferation-inducing ligand (APRIL), cytotoxicT lymphocyte-associated protein-4-inducible costimulator (CTLA4-ICOS) y recombination activating gene (RAG1), polimorfismo que podría asociar un mayor riesgo de presentar DSIgA, enfermedad celíaca e IDCV2,4,6–10. Aparte de compartir defectos genéticos, la agregación familiar y el conocido riesgo de que el DSIgA progrese hacia una IDCV, sugieren que es probable que formen parte de un amplio espectro de la misma enfermedad2,11. Además, ciertos haplotipos HLA (A1, A29, B8, B14, DR3, DQ2) se han asociado a una mayor susceptibilidad de presentar DSIgA, aunque esta asociación no está clara ya que estos haplotipos se asocian también a enfermedades autoinmunes, frecuentemente asociadas a este defecto2,12–14.
La mayoría de los pacientes (85-90%) afectados de DSIgA son asintomáticos. La clínica que presentan los pacientes sintomáticos presenta un rango muy amplio que incluye infecciones sinopulmonares recurrentes, infecciones y trastornos gastrointestinales, alergias, procesos autoinmunes y neoplasias con mayor frecuencia que la población general2,5,15,16.
Aunque la mayoría de los casos se diagnostican por determinación de las concentraciones séricas de inmunoglobulinas dentro del estudio inmunitario por infecciones respiratorias de repetición, muchos otros casos se diagnostican «accidentalmente», como hallazgos dentro de una evaluación de laboratorio en enfermedad celíaca, alergias o enfermedades autoinmunes8.
Debido a la alta prevalencia descrita entre familiares, y a que no se han encontrado estudios que comparen las diferencias clínicas y/o inmunológicas entre los casos familiares (CF) y los esporádicos (CE) para valorar la gravedad, el pronóstico y la necesidad de realizar otras pruebas complementarias, el objetivo de nuestro estudio fue evaluar la prevalencia de DSIgA en FPG de pacientes con DSIgA, determinar si el cribado en miembros familiares puede ser una herramienta útil en el manejo clínico de esta patología y comparar las características clínicas e inmunológicas en niños diagnosticados de DSIgA esporádico con aquellos casos en los que existía algún familiar de primer grado (FPG) afectado.
Pacientes y métodosPoblación de estudioSe realizó un estudio transversal descriptivo donde se incluyó a todos los pacientes con DSIgA y sus FPG diagnosticados y/o seguidos en la Unidad de Patología Infecciosa e Inmunodeficiencias de Pediatría del Hospital Universitari Vall d’Hebron de Barcelona que cumplieron los criterios de inclusión desde octubre del 2010 hasta septiembre del 2011. Estos pacientes fueron diagnosticados, o por presentar clínica o de forma incidental, al estudiar otras patologías, como enfermedades autoinmunitarias o alérgicas.
Protocolo de estudioSe cumplimentó un cuestionario para todos los participantes que incluía datos demográficos (edad, sexo, consanguinidad) y clínicos (infecciones, alergias, autoinmunidad, patología respiratoria, gastrointestinal, dermatológica y neoplasias), todos recogidos por un mismo investigador. Para el estudio de los casos se extrajeron muestras sanguíneas para el análisis de hemograma, inmunoglobulinas y subclases de IgG séricas, anticuerpos antinucleares (ANA), isohemaglutininas, antiestreptolisina O (ASLO) y respuesta vacunal frente a tétanos, sarampión, parotiditis, rubéola, Haemophilus influenzae (H. influenzae) b y Streptococcus pneumoniae (S. pneumoniae). En el análisis de los familiares, se incluyó el hemograma e inmunoglobulinas (IgG, IgA e IgM). Este estudio fue aprobado por el comité ético del hospital.
Criterios inclusión y definicionesSe incluyó a aquellos pacientes pediátricos con diagnóstico definitivo de DSIgA según los criterios de la ESID y PAGID17, entre 4 y 16 años, y a sus FPG, cuya familia al completo aceptó participar y firmó el consentimiento informado.
Así, se definió como DSIgA aquella situación en que los niveles de IgA en sangre eran inferiores a 7mg/dl (0,07g/l) con niveles normales de IgG e IgM y una respuesta a anticuerpos normal en mayores de 4 años de edad17.
Se definió como CF a aquel paciente diagnosticado o seguido en nuestra unidad que, al estudiar a sus familiares, se comprobó que había más de un miembro de la misma familia afectado. Si solo estaba afectado el caso índice, se consideró como CE.
Los familiares de primer grado con DSIgA se denominaron FPG enfermos (FPG-E) y aquellos familiares a los que no se les detectó DSIgA se clasificaron como FPG sanos (FPG-S).
Estudios de laboratorioLos parámetros hematológicos y bioquímicos fueron obtenidos mediante métodos de rutina del laboratorio. Las inmunoglobulinas (Ig) fueron determinadas mediante nefelometría, las subclases de IgG mediante nefelometría con anticuerpos monoclonales (MoAb) y la respuesta vacunal en aquellos inmunizados a través de métodos serológicos convencionales.
Se describieron como concentraciones séricas de inmunoglobulinas (IgM, IgG, IgA, IgE), subclases de IgG, isohemaglutininas y antiestreptolisinas descendidas cuando al menos estaban por debajo de 2 desviaciones estándar (DE) de la media de los valores de referencia marcados por Jollif et al.18 para el sujeto en función de la edad. Se consideraron como negativos unos valores de ASLO<200 UI/ml y de isohemaglutininas menores de 1/32, tanto para IgM como para IgG. La respuesta vacunal fue considerada como negativa en aquellos con confirmación previa de la vacunación correspondiente y unos valores por debajo del punto de corte de nuestro centro. Se consideraron patológicos los valores de ANA cuando fueron mayores de 1/80.
Análisis estadísticoPara el análisis de los datos cualitativos se usó el test exacto de Fisher y para las variables cuantitativas la t de Student con un 5% de significación. Para el análisis bivariante se calcularon odds ratio (OR). El análisis estadístico se llevó a cabo mediante el programa SPSS (SPSS Statistics for Windows, Versión 17.0, SPSS Inc, Chicago, EE. UU.).
ResultadosCaracterísticas sociodemográficasEl tamaño de muestra fue de 130 participantes, 42 casos y 88 FPG (fig. 1).

La edad media ± DE de los casos con DSIgA fue de 11 años ± 4,54 y un 57% eran de sexo femenino sin encontrar diferencias entre los CF y CE (p=0,17 y 0,10 respectivamente). La edad media de los FPG fue de 34±15,85 años y un 57% (50) eran de sexo femenino No se recogió ningún caso de consanguinidad.
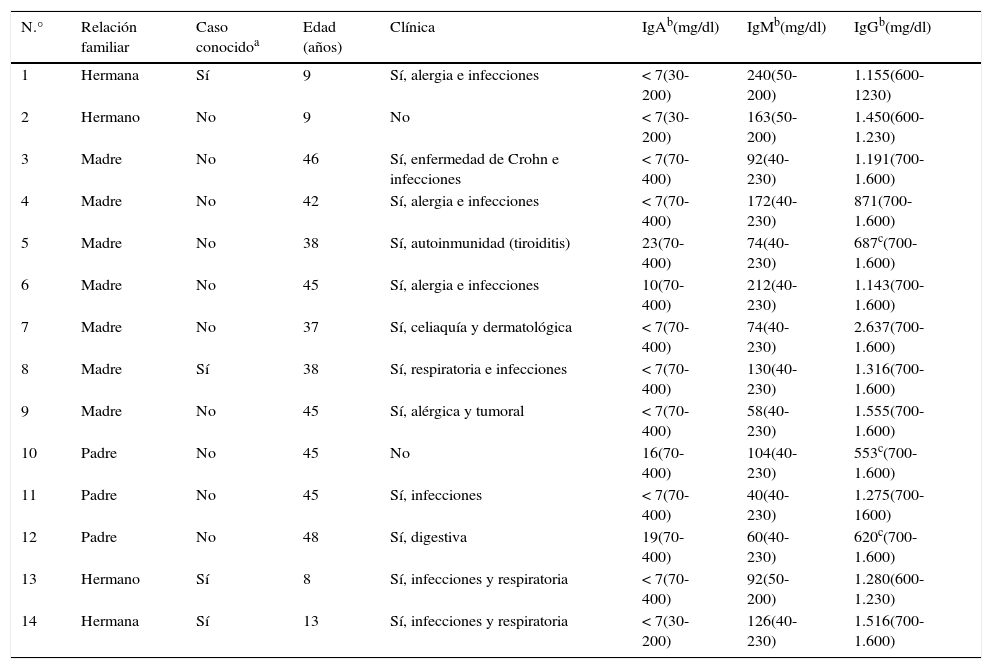
Casos familiaresEn 13 de los 42 pacientes con DSIgA registrados (31%) existía uno o más casos entre sus familiares. La frecuencia de DSIgA en los FPG fue estimada en un caso por cada 6 miembros familiares. Tenían un diagnóstico conocido previo de DSIgA una madre, un hermano y 2 hermanas (tabla 1).
Características sociodemográficas, clínicas y de laboratorio de los familiares de primer grado enfermos (FPG-E)
| N.° | Relación familiar | Caso conocidoa | Edad (años) | Clínica | IgAb(mg/dl) | IgMb(mg/dl) | IgGb(mg/dl) |
|---|---|---|---|---|---|---|---|
| 1 | Hermana | Sí | 9 | Sí, alergia e infecciones | < 7(30-200) | 240(50-200) | 1.155(600-1230) |
| 2 | Hermano | No | 9 | No | < 7(30-200) | 163(50-200) | 1.450(600-1.230) |
| 3 | Madre | No | 46 | Sí, enfermedad de Crohn e infecciones | < 7(70-400) | 92(40-230) | 1.191(700-1.600) |
| 4 | Madre | No | 42 | Sí, alergia e infecciones | < 7(70-400) | 172(40-230) | 871(700-1.600) |
| 5 | Madre | No | 38 | Sí, autoinmunidad (tiroiditis) | 23(70-400) | 74(40-230) | 687c(700-1.600) |
| 6 | Madre | No | 45 | Sí, alergia e infecciones | 10(70-400) | 212(40-230) | 1.143(700-1.600) |
| 7 | Madre | No | 37 | Sí, celiaquía y dermatológica | < 7(70-400) | 74(40-230) | 2.637(700-1.600) |
| 8 | Madre | Sí | 38 | Sí, respiratoria e infecciones | < 7(70-400) | 130(40-230) | 1.316(700-1.600) |
| 9 | Madre | No | 45 | Sí, alérgica y tumoral | < 7(70-400) | 58(40-230) | 1.555(700-1.600) |
| 10 | Padre | No | 45 | No | 16(70-400) | 104(40-230) | 553c(700-1.600) |
| 11 | Padre | No | 45 | Sí, infecciones | < 7(70-400) | 40(40-230) | 1.275(700-1600) |
| 12 | Padre | No | 48 | Sí, digestiva | 19(70-400) | 60(40-230) | 620c(700-1.600) |
| 13 | Hermano | Sí | 8 | Sí, infecciones y respiratoria | < 7(70-400) | 92(50-200) | 1.280(600-1.230) |
| 14 | Hermana | Sí | 13 | Sí, infecciones y respiratoria | < 7(30-200) | 126(40-230) | 1.516(700-1.600) |
De los casos familiares diagnosticados, un 50% (7) fueron madres, un 21,4% (3) padres, 14,3% (2) hermanos y 14,3% (2) hermanas (p=0,55; OR=1,96; IC del 95%, 0,62-6,21). En otras palabras, un 22% de las madres, un 14,5% de los hermanos y hermanas y un 10% de los padres mostraron un DSIgA. Las características sociodemográficas, clínicas y de laboratorio de cada FPG-E por separado se muestran en la tabla 1. Tomando una prevalencia estimada de DSIgA de 1/600 en caucásicos como representación de nuestra muestra, el número necesario a analizar calculado para encontrar un CF con DSIgA fue de 6 familiares.
Manifestaciones clínicasTodos los niños con DSIgA presentaron algún tipo de manifestación clínica. Las infecciones registradas se muestran en la tabla 2. Solo en 2 (6,9%) pacientes de CE con diarrea se aislaron parásitos.
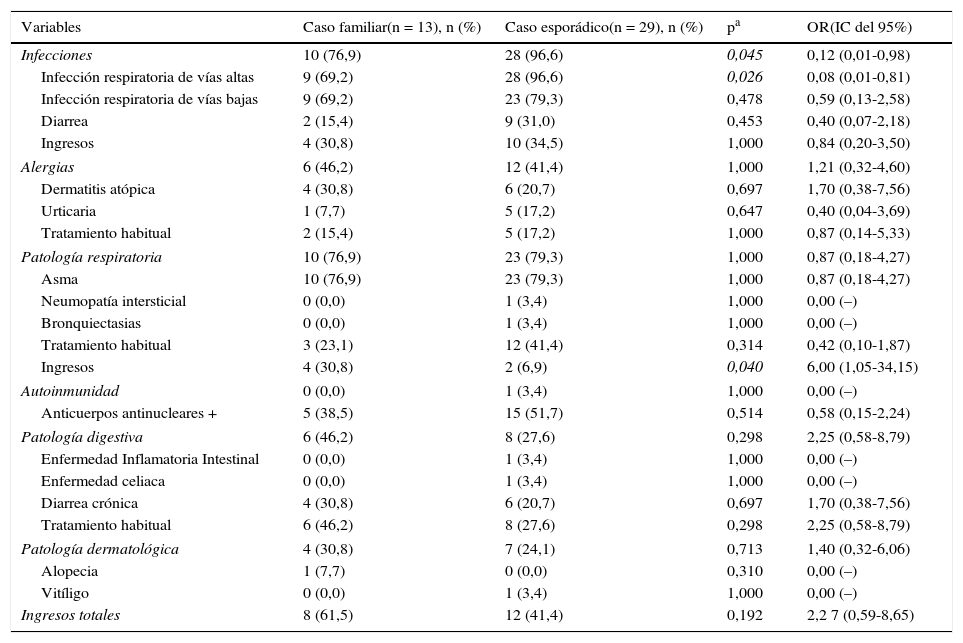
Características clínicas de los CF y CE
| Variables | Caso familiar(n = 13), n (%) | Caso esporádico(n = 29), n (%) | pa | OR(IC del 95%) |
|---|---|---|---|---|
| Infecciones | 10 (76,9) | 28 (96,6) | 0,045 | 0,12 (0,01-0,98) |
| Infección respiratoria de vías altas | 9 (69,2) | 28 (96,6) | 0,026 | 0,08 (0,01-0,81) |
| Infección respiratoria de vías bajas | 9 (69,2) | 23 (79,3) | 0,478 | 0,59 (0,13-2,58) |
| Diarrea | 2 (15,4) | 9 (31,0) | 0,453 | 0,40 (0,07-2,18) |
| Ingresos | 4 (30,8) | 10 (34,5) | 1,000 | 0,84 (0,20-3,50) |
| Alergias | 6 (46,2) | 12 (41,4) | 1,000 | 1,21 (0,32-4,60) |
| Dermatitis atópica | 4 (30,8) | 6 (20,7) | 0,697 | 1,70 (0,38-7,56) |
| Urticaria | 1 (7,7) | 5 (17,2) | 0,647 | 0,40 (0,04-3,69) |
| Tratamiento habitual | 2 (15,4) | 5 (17,2) | 1,000 | 0,87 (0,14-5,33) |
| Patología respiratoria | 10 (76,9) | 23 (79,3) | 1,000 | 0,87 (0,18-4,27) |
| Asma | 10 (76,9) | 23 (79,3) | 1,000 | 0,87 (0,18-4,27) |
| Neumopatía intersticial | 0 (0,0) | 1 (3,4) | 1,000 | 0,00 (–) |
| Bronquiectasias | 0 (0,0) | 1 (3,4) | 1,000 | 0,00 (–) |
| Tratamiento habitual | 3 (23,1) | 12 (41,4) | 0,314 | 0,42 (0,10-1,87) |
| Ingresos | 4 (30,8) | 2 (6,9) | 0,040 | 6,00 (1,05-34,15) |
| Autoinmunidad | 0 (0,0) | 1 (3,4) | 1,000 | 0,00 (–) |
| Anticuerpos antinucleares + | 5 (38,5) | 15 (51,7) | 0,514 | 0,58 (0,15-2,24) |
| Patología digestiva | 6 (46,2) | 8 (27,6) | 0,298 | 2,25 (0,58-8,79) |
| Enfermedad Inflamatoria Intestinal | 0 (0,0) | 1 (3,4) | 1,000 | 0,00 (–) |
| Enfermedad celiaca | 0 (0,0) | 1 (3,4) | 1,000 | 0,00 (–) |
| Diarrea crónica | 4 (30,8) | 6 (20,7) | 0,697 | 1,70 (0,38-7,56) |
| Tratamiento habitual | 6 (46,2) | 8 (27,6) | 0,298 | 2,25 (0,58-8,79) |
| Patología dermatológica | 4 (30,8) | 7 (24,1) | 0,713 | 1,40 (0,32-6,06) |
| Alopecia | 1 (7,7) | 0 (0,0) | 0,310 | 0,00 (–) |
| Vitíligo | 0 (0,0) | 1 (3,4) | 1,000 | 0,00 (–) |
| Ingresos totales | 8 (61,5) | 12 (41,4) | 0,192 | 2,2 7 (0,59-8,65) |
CE: casos esporádicos; CF: casos familiares; IC: intervalo de confianza; OR: odds ratio.
Con respecto a fenómenos de autoinmunidad, tan solo se describió un caso de tiroiditis de Hashimoto. No se contabilizó ninguna neoplasia.
No se encontraron diferencias clínicas significativas entre los CF y los CE. De hecho, incluso de las infecciones fueron más frecuentes en CE (p=0,045, OR=0,12; IC del 95%, 0,01-0,98). Por el contrario, es cierto que los CF presentaron más ingresos de causa respiratoria que los CE (p=0,04, OR=6, IC del 95%, 1,05-34,15), dato que no se confirma al evaluar el total de ingresos (tabla 2).
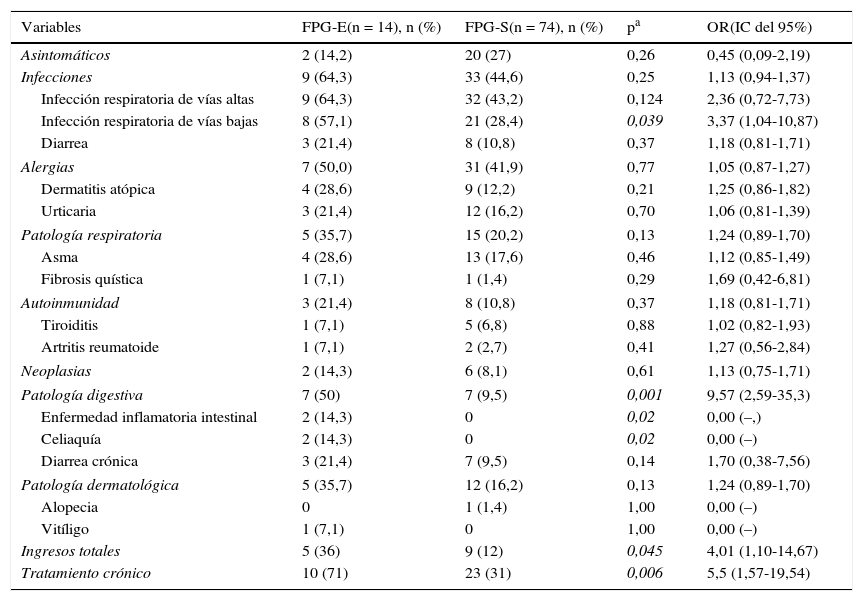
Cuando se evaluaron los FPG, no se encontraron diferencias en cuanto a manifestaciones clínicas entre ambos grupos a excepción de la presencia de patología digestiva, significativamente más frecuente en los FPG-E (tabla 3). Tenían psoriasis 10 FPG (13,5%), todos del grupo FPG-S. Todas las neoplasias reportadas fueron tumores sólidos sin diferencias entre ambos grupos.
Características clínicas de los FPG-S y los FPG-E
| Variables | FPG-E(n = 14), n (%) | FPG-S(n = 74), n (%) | pa | OR(IC del 95%) |
|---|---|---|---|---|
| Asintomáticos | 2 (14,2) | 20 (27) | 0,26 | 0,45 (0,09-2,19) |
| Infecciones | 9 (64,3) | 33 (44,6) | 0,25 | 1,13 (0,94-1,37) |
| Infección respiratoria de vías altas | 9 (64,3) | 32 (43,2) | 0,124 | 2,36 (0,72-7,73) |
| Infección respiratoria de vías bajas | 8 (57,1) | 21 (28,4) | 0,039 | 3,37 (1,04-10,87) |
| Diarrea | 3 (21,4) | 8 (10,8) | 0,37 | 1,18 (0,81-1,71) |
| Alergias | 7 (50,0) | 31 (41,9) | 0,77 | 1,05 (0,87-1,27) |
| Dermatitis atópica | 4 (28,6) | 9 (12,2) | 0,21 | 1,25 (0,86-1,82) |
| Urticaria | 3 (21,4) | 12 (16,2) | 0,70 | 1,06 (0,81-1,39) |
| Patología respiratoria | 5 (35,7) | 15 (20,2) | 0,13 | 1,24 (0,89-1,70) |
| Asma | 4 (28,6) | 13 (17,6) | 0,46 | 1,12 (0,85-1,49) |
| Fibrosis quística | 1 (7,1) | 1 (1,4) | 0,29 | 1,69 (0,42-6,81) |
| Autoinmunidad | 3 (21,4) | 8 (10,8) | 0,37 | 1,18 (0,81-1,71) |
| Tiroiditis | 1 (7,1) | 5 (6,8) | 0,88 | 1,02 (0,82-1,93) |
| Artritis reumatoide | 1 (7,1) | 2 (2,7) | 0,41 | 1,27 (0,56-2,84) |
| Neoplasias | 2 (14,3) | 6 (8,1) | 0,61 | 1,13 (0,75-1,71) |
| Patología digestiva | 7 (50) | 7 (9,5) | 0,001 | 9,57 (2,59-35,3) |
| Enfermedad inflamatoria intestinal | 2 (14,3) | 0 | 0,02 | 0,00 (–,) |
| Celiaquía | 2 (14,3) | 0 | 0,02 | 0,00 (–) |
| Diarrea crónica | 3 (21,4) | 7 (9,5) | 0,14 | 1,70 (0,38-7,56) |
| Patología dermatológica | 5 (35,7) | 12 (16,2) | 0,13 | 1,24 (0,89-1,70) |
| Alopecia | 0 | 1 (1,4) | 1,00 | 0,00 (–) |
| Vitíligo | 1 (7,1) | 0 | 1,00 | 0,00 (–) |
| Ingresos totales | 5 (36) | 9 (12) | 0,045 | 4,01 (1,10-14,67) |
| Tratamiento crónico | 10 (71) | 23 (31) | 0,006 | 5,5 (1,57-19,54) |
FPG-E: familiares de primer grado enfermos; FPG-S: de primer grado sanos; IC: intervalo de confianza; OR: odds ratio.
Como marcador de gravedad clínica se valoró la necesidad de ingresos hospitalarios y de tratamiento crónico. Se demostró una proporción significativamente mayor de ingresos totales en los FPG-E con respecto a los FPG-S teniendo un riesgo aumentado de ingresar globalmente 4 veces mayor. Los motivos de ingreso en los FPG-E fueron por causas infecciosas (2), respiratorias (2) y un caso secundario a patología digestiva. En los FPG-S, 8 de los 9 ingresos fueron causados por infecciones y el restante a patología digestiva. Además, los FPG-E necesitaban tratamiento crónico 5 veces más que los FPG-S (tabla 3). En los FPG-E, la mayoría (7) se debía a patología digestiva y el resto recibía tratamiento crónico frente a patología respiratoria (2) y alérgica (1). El tratamiento habitual que se reportó en los FPG-S fue en un 12 (52%) secundario a alteraciones gastrointestinales, 7 (31%) por patología respiratoria y 4 (17%) por alergias.
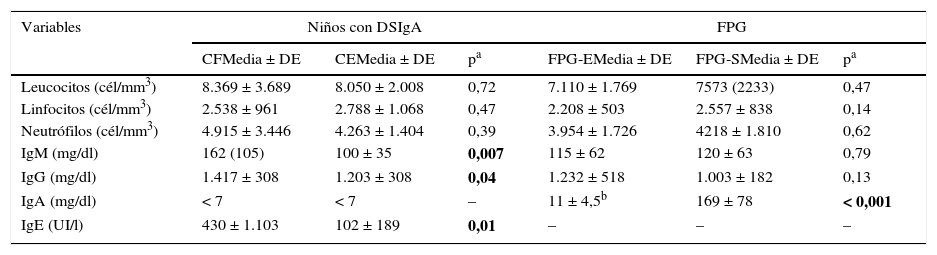
Estudios de laboratorioLos niveles de inmunoglobulinas, leucocitos totales, linfocitos y neutrófilos de los casos y los FPG se exponen en la tabla 4. Las cifras de IgG, IgM e IgE fueron significativamente mayores en los CF que en los CE (p=0,007, p = 0,04 y p = 0,01, respectivamente). Un paciente de cada grupo presentó concentraciones de subclases de IgG descendidas (ambas IgG2). La respuesta humoral fue muy similar en ambos grupos y se describe en la tabla 5.
Parámetros del hemograma y concentraciones séricas de inmunoglobulinas en CF vs. CE y FPG-S vs. FPG-E
| Variables | Niños con DSIgA | FPG | ||||
|---|---|---|---|---|---|---|
| CFMedia ± DE | CEMedia ± DE | pa | FPG-EMedia ± DE | FPG-SMedia ± DE | pa | |
| Leucocitos (cél/mm3) | 8.369 ± 3.689 | 8.050 ± 2.008 | 0,72 | 7.110 ± 1.769 | 7573 (2233) | 0,47 |
| Linfocitos (cél/mm3) | 2.538 ± 961 | 2.788 ± 1.068 | 0,47 | 2.208 ± 503 | 2.557 ± 838 | 0,14 |
| Neutrófilos (cél/mm3) | 4.915 ± 3.446 | 4.263 ± 1.404 | 0,39 | 3.954 ± 1.726 | 4218 ± 1.810 | 0,62 |
| IgM (mg/dl) | 162 (105) | 100 ± 35 | 0,007 | 115 ± 62 | 120 ± 63 | 0,79 |
| IgG (mg/dl) | 1.417 ± 308 | 1.203 ± 308 | 0,04 | 1.232 ± 518 | 1.003 ± 182 | 0,13 |
| IgA (mg/dl) | < 7 | < 7 | – | 11 ± 4,5b | 169 ± 78 | < 0,001 |
| IgE (UI/l) | 430 ± 1.103 | 102 ± 189 | 0,01 | – | – | – |
CE: casos esporádicos; CF: casos familiares; DE: desviación estándar; DSIgA: déficit selectivo de IgA; FPG: familiares de primer grado; FPG-E: FPG enfermos; FPG-S: FPG sanos.
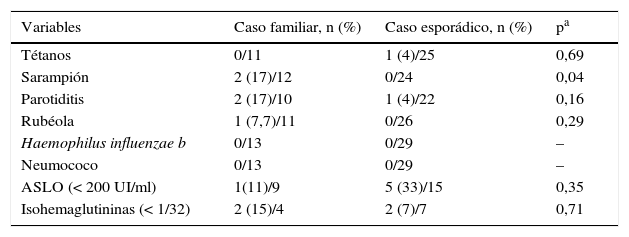
Estudio de producción de anticuerpos en los casos familiares y esporádicos
| Variables | Caso familiar, n (%) | Caso esporádico, n (%) | pa |
|---|---|---|---|
| Tétanos | 0/11 | 1 (4)/25 | 0,69 |
| Sarampión | 2 (17)/12 | 0/24 | 0,04 |
| Parotiditis | 2 (17)/10 | 1 (4)/22 | 0,16 |
| Rubéola | 1 (7,7)/11 | 0/26 | 0,29 |
| Haemophilus influenzae b | 0/13 | 0/29 | – |
| Neumococo | 0/13 | 0/29 | – |
| ASLO (< 200 UI/ml) | 1(11)/9 | 5 (33)/15 | 0,35 |
| Isohemaglutininas (< 1/32) | 2 (15)/4 | 2 (7)/7 | 0,71 |
A pesar de no existir diferencias en los niveles de IgG e IgM entre ambos grupos de FPG, se detectaron 3 casos de hipogammaglobulinemia G en los FPG-E y ninguno en los de FPG-S (tabla 4).
DiscusiónNo se ha podido demostrar un fenotipo clínico más grave en los casos familiares de DSIgA de nuestra serie. Tan solo pudimos demostrar un riesgo claramente mayor de ingreso por patología respiratoria (la mayoría por asma) en los CF que podría indicar un fenotipo más grave, pero este punto no se confirmó al evaluar globalmente los ingresos por cualquier causa en ambos grupos.
De todos modos, hay una serie de consideraciones que debemos tener presentes. Así, aunque en la mayoría de las series reportadas en la literatura los pacientes con DSIgA son asintomáticos2,19, en nuestro estudio todos los pacientes reportaron algún tipo de clínica, posiblemente debido a un sesgo de selección de la muestra por el centro en el que se controlaban. Este hecho puede haber contribuido a la ausencia de diferencias clínicas significativas en nuestra muestra. Por otra parte, el ser un estudio retrospectivo implica un cierto riesgo de pérdida de información, sobre todo en cuanto a manifestaciones clínicas banales, como infecciones leves.
No se encontraron diferencias en la distribución del tipo de infecciones respecto a lo reportado en la literatura5,16 y, a pesar de que globalmente las infecciones fueron más frecuentes en los CE, no hubo diferencias en cuanto a la necesidad de ingreso hospitalario, lo que indica un curso leve en ambos grupos. La baja prevalencia de procesos alérgicos y patología intestinal, autoinmune y tumoral de nuestra muestra en comparación con otras series1,2,15,16,19–24 —probablemente en relación con el hecho de tratarse de una población pediátrica— impiden llegar a conclusiones respecto al papel que pueda desempeñar la agregación familiar como factor de riesgo de desarrollar este tipo de patología. No obstante, fue llamativa la alta prevalencia de ANA positivos en pacientes de ambos grupos sin manifestaciones autoinmunes evidentes, tal vez relacionado con el DSIgA como epifenómeno de los trastornos autoinmunes, capaz de conferir una mayor probabilidad de desarrollar un trastorno autoinmune en la edad adulta que la población general o como parte de una respuesta compensatoria frente a infecciones y frente a un constante estado inflamatorio del organismo como indican Fusaro et al.25. Del mismo modo, los valores de IgE total también fueron superiores en los CF a pesar de no presentar una mayor prevalencia de procesos alérgicos clínicamente significativos.
Las concentraciones séricas de IgG e IgM fueron significativamente mayores en los CF sin que se pudiera establecer una causa clara para este hallazgo, ya que no se constató en este grupo mayor prevalencia o gravedad de procesos infecciosos o inflamatorios que pudieran justificarlo.
En cuanto a la calidad de la respuesta humoral, medida a través de isohemaglutininas y títulos de ASLO, han sido deficientes en un número no despreciable de la muestra (3 en CF y 6 en CE), por ello sería conveniente realizar estas determinaciones en todos los casos. La respuesta a vacunas incluidas en nuestro calendario vacunal (tétanos, sarampión, parotiditis, rubéola, H. influenzae y S. pneumoniae) ha sido buena en ambos grupos, con diferencia de la vacuna del sarampión, donde los únicos casos reportados de mala respuesta pertenecían a CF. Independientemente del grupo al que pertenezcan, consideramos imprescindible un seguimiento estricto de estos pacientes a lo largo de la evolución para valorar si estos hallazgos pueden ser premonitorios del desarrollo de inmunodeficiencias clínicamente más significativas como la IDCV coincidiendo con otras publicaciones11.
En cuanto a la utilidad del estudio en familiares, parece razonable afirmar que los pacientes detectados a partir del cribado familiar de los pacientes pediátricos con DSIgA presentan una sintomatología clínica suficientemente significativa como para justificar este cribado, más teniendo en cuenta la elevada prevalencia de casos familiares demostrada en el presente estudio y en otras publicaciones3,26,27.
Así, el factor de riesgo más significativo para el desarrollo de DSIgA demostrado en estudios previos es la existencia de una historia familiar positiva28. Debido a las diferencias genéticas demostradas según la etnia, es de esperar que existan tasas diferentes de familiares afectados en distintas regiones del mundo3. En nuestro análisis, la frecuencia de algún FPG afectado fue de un 16%, siendo diagnosticados de novo más de un 70% de los familiares. Existía más de un caso en una misma familia en un 31%. Este porcentaje es similar a lo publicado en otras series, que estiman cifras de hasta un 32,4% con una elevada prevalencia de consanguinidad inexistente en nuestra muestra3 y bastante más elevadas que las medidas en la población general, con prevalencias en caucásicos de un caso por cada 600.
De acuerdo con los resultados de nuestro estudio, el riesgo de tener DSIgA en FPG de pacientes con esta patología fue ligeramente mayor en las madres (22%) que en padres (10%) y hermanos (14,5%), aunque sin encontrar grandes diferencias. Estos datos no están en consonancia con lo publicado en estudios previos, donde encuentran un riesgo mayor en hermanos3,27,28. Vorechovský et al.28 analizaron las concentraciones séricas de Ig de familiares cercanos de pacientes suecos con DSIgA e IDCV estimando un riesgo relativo para los hermanos de pacientes con DSIgA en aproximadamente 50. Sin embargo, se ha demostrado una diferente penetrancia entre ambos progenitores en la transmisión de DSIgA a los hijos29. Algunas investigaciones han encontrado una mayor proporción de niños con DSIgA nacidos de madres con esta patología que aquellos nacidos de padres con el mismo defecto28,30. Este tipo de herencia con más predominancia en la transmisión materna podría relacionarse con defectos mitocondriales y quizá podría explicar en parte el mayor porcentaje de madres afectas encontradas en nuestro estudio, ya que el número de hermanos (un hermano/a por cada madre) es pequeño en comparación con otros publicados3,27.
A pesar de que en nuestro estudio solo un 14% de los FPG-E estaban asintomáticos, este hecho no se diferenciaba de los FPG-S, por lo que es de suponer que las patologías referidas en los FPG-E eran similares a las de la población general. En 2008, Aghamohammadi et al.27 presentó un estudio que afirmaba que todos los familiares de pacientes con DSIgA que también presentaban este defecto estaban asintomáticos; sin embargo, no los comparó con la cohorte de familiares sanos, hecho que podría justificar nuestros resultados. En un estudio reciente de casos-controles donde evaluaron los síntomas de adultos con DSIgA frente a personas sanas hallaron mayores tasas de infecciones, fenómenos alérgicos y autoinmunidad en los pacientes con DSIgA26, hecho que no se confirma en nuestra serie. En cambio, si tomamos como marcador de gravedad clínica los ingresos hospitalarios y la necesidad de recibir un tratamiento crónico, sí se confirmaría la afirmación realizada por Jorgensen et al.26.
A pesar de no haber podido demostrar unos valores significativamente menores de IgG e IgM, como indicaban otros autores que los proponían, como marcadores de riesgo de evolución a IDCV3,27, sí se ha a detectado 3 pacientes adultos con hipogammaglobulinemia IgG que deberán ser estudiados en profundidad. En este sentido, la no evaluación de las subclases de IgG y la producción de anticuerpos en los familiares suponen una limitación a la hora de la validar estos hallazgos.
En conclusión, consideramos que la elevada prevalencia de DSIgA en FPG de pacientes con esta patología asociada a una mayor gravedad clínica, medida de forma indirecta a través de los ingresos y la necesidad de tratamiento crónico, hacen pensar que los FPG podrían beneficiarse de programas de cribado, sobre todo si tenemos en cuenta que esta medida podría ser coste-efectiva por el bajo número de familiares que necesitan ser analizados para encontrar un caso positivo. De esta manera podrían iniciar, en caso de que lo necesitasen, profilaxis o tratamientos que evitasen un mayor número de ingresos, o simplemente realizar un seguimiento periódico por el riesgo de progresión a IDCV.
AutoríaP. Soler-Palacín y E. Cobos-Carrascosa han contribuido por igual en el estudio.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
