

Selective immunoglobulin A deficiency (SIgAD), the most common primary immunodeficiency, is often asymptomatic. High rates of familial clustering have been described in SIgAD, but the causative genetic defect and mechanism of inheritance are unknown.
ObjectivesTo determine whether familial SIgAD cases show more severe clinical and immunological characteristics than sporadic ones; to investigate the utility of screening first-degree relatives (FDRs) of these patients, and to determine whether symptoms in affected family members are important enough to justify screening.
Patients and methodsDescriptive, cross-sectional study (October 2010–September 2011) of all patients with SIgAD followed up in our centre. Demographic, clinical, and analytical data were reviewed. A familial case was defined as an SIgAD patient with at least 1 affected FDR.
ResultsOf the 130 participants, 42 were SIgAD patients and 88 FDR. There were 13 (31%) familial cases and 14 (16%) affected FDRs. Six family members had to be studied in order to detect 1 affected member. There were no clinical differences between familial and sporadic SIgAD cases. The percentages of intestinal disease (p=0.001, OR=9.57, 95% CI 2.59–35.3), hospitalisations (p=0.045, OR=4.01; 95% CI 1.10–14.67], and need for chronic treatment (p=0.006, OR=5.5; 95% CI 1.57–19.54) were higher in affected FDRs than in unaffected ones.
ConclusionsThe symptoms were not more severe in familial than sporadic SIgAD cases. Nonetheless, the elevated prevalence of affected FDRs with significant morbidity may justify routine screening of close family members of these patients.
El déficit selectivo de IgA (DSIgA) es la inmunodeficiencia primaria más frecuente, siendo a menudo asintomática. Se ha descrito una elevada agregación familiar, sin conocerse el defecto genético causante ni su mecanismo hereditario.
ObjetivosDefinir la utilidad del cribado de los familiares de primer grado de los pacientes con DSIgA valorando si los casos familiares presentan unas características clínicas e inmunológicas más graves que los casos esporádicos (CE) y si los familiares diagnosticados de DSIgA presentan sintomatología clínica significativa para justificar su cribado.
Pacientes y métodosEstudio transversal descriptivo (octubre del 2010-septiembre del 2011) de todos los pacientes con DSIgA controlados en nuestro centro, con revisión de datos demográficos, clínicos y analíticos. Se consideró como caso familiar (CF) todo aquel con al menos un familiar de primer grado (FPG) con DSIgA.
ResultadosDe los 130 participantes, 42 eran pacientes con DSIgA y 88 FPG. Se diagnosticaron 13 CF (31%), 29 CE (69%) y 14 (16%) FPG enfermos (FPG-E). El número necesario a analizar para encontrar un FPG-E fue de 6 familiares. No hubo diferencias clínicas entre los pacientes. Hubo una proporción mayor de patología intestinal (p=0,001, OR=9,57, IC del 95%, 2,59–35,3), ingresos (p=0,045, OR=4,01; IC del 95%, 1,10–14,67) y necesidad de tratamiento crónico (p=0,006, OR=5,5; IC del 95%, 1,57–19,54) en los FPG-E con respecto a los FPG sanos.
ConclusionesA pesar de no encontrar más complicaciones clínicas en los CF de DSIgA, la elevada prevalencia de familiares afectados con afectación clínica significativa podría justificar la realización sistemática de estos programas de cribado.
Selective IgA deficit (SIgAD) belongs to the group of primary immunodeficiencies characterised by a deficit in the production of antibodies. Although SIgAD is the most frequent primary immunodeficiency (PID), its prevalence may vary from 1/163 to 1/18,500 cases, based on the studied population and the definition of SIgAD.1,2 These variations support a genetic predisposition, and new cases can appear sporadically or be transmitted in a dominant autosomal or recessive manner, or through polygenic inheritance. Thus, a 30% probability of SIgAD has been estimated if there is an affected relative,3 but no specific genetic defect has been identified, nor has a clear Mendelian inheritance pattern been proven, and expression and penetrance differences persist.2,4,5 Several genes potentially implicated in its pathogenesis have been identified, suggesting that SIgAD is probably a heterogeneous group of diseases and/or genetic anomalies similar to common variable immunodeficiency (CVID), notably transmembrane activator and calcium modulator (TACI), B-cell activating factor (BAFF-receptor), proliferation-inducing ligand (APRIL), cytotoxic T lymphocyte-associated protein-4-inducible co-stimulator (CTLA4-ICOS) and recombinant activating gene (RAG1), polymorphism, which could be associated a higher risk of presenting SIgAD, coeliac disease, and CVID.2,4,6–10 In addition to sharing genetic defects, familial aggregation and the known risk of SIgAD progressing to CVID suggest that they are probably part of a wide spectrum of the same disease.2,11 Furthermore, certain haplotypes HLA (A1, A29, B8, B14, DR3, DQ2) have been associated with a greater risk of SIgAD, although this association is not clear because these haplotypes are also associated with the autoimmune diseases frequently associated with this defect.2,12–14
Most patients (85%–90%) affected by SIgAD are asymptomatic. The clinical signs of symptomatic patients are wide ranging, including recurrent sinopulmonary infections, infections and gastrointestinal disorders, allergies, autoimmune processes and neoplasias, with more frequency than the general population.2,5,15,16
Although the diagnosis of SIgAD is usually made by analysis of serum immunoglobulins during the evaluation of recurrent respiratory infections, many other cases are diagnosed “accidentally”, as part of a laboratory evaluation for coeliac disease, allergy, or autoimmune disease.8
Prevalence of SIgAD is high among family members, and no studies have as yet been undertaken to compare the clinical and/or immunological difference among familial (FC) and sporadic (SC) cases. As a result, the severity, prognosis and need for complementary tests cannot be assessed. In this context, we set out to evaluate the prevalence of SIgAD in first-degree relatives (FDR) of SIgAD patients, determine whether screening family members is a useful strategy in the clinical management of this pathology, and compare the clinical and immunological characteristics in children diagnosed with sporadical SIgAD with those cases where there is an affected first-degree relative (FDR).
Patients and methodsStudy populationA descriptive cross-sectional study was carried out from October 2010 to September 2011. All patients with SIgAD and their FDRs diagnosed and/or followed-up at the Paediatric Unit of Infectious Pathology and Immunodeficiencies of the Hospital Universitari Vall d’Hebron in Barcelona who met inclusion criteria were selected for the study. These patients were diagnosed, either because they had clinical signs or incidentally when studying other pathologies, such as allergic or autoimmune diseases.
Study protocolA questionnaire was administered to all participants by the same researcher. The information collected included demographic data (age, sex, consanguinity) and clinical signs (infections, allergies, autoimmunity, respiratory, gastrointestinal, dermatologic pathology and neoplasia), blood samples were collected for complete blood count, study of immunoglobulins and subclasses of serum IgG, antinuclear antibodies (ANA), isohaemaglutinines, antistreptolysin O (ASLO) and vaccine response against tetanus, measles, mumps, rubella, Haemophilus influenzae (H. influenzae) b and Streptococcus pneumoniae (S. pneumoniae). In family screening, complete blood count and immunoglobulins were included (IgG, IgA and IgM). This study was approved by the ethics committee of the hospital.
Inclusion criteria and definitionsPaediatric patients aged between 4 and 8 years with final diagnosis of SIgAD according to the criteria of the ESID and PAGID17 were included together with their FDRs, whose families all agreed to participate and signed the informed consent form.
SIgAD was defined as a blood IgA level below 7mg/dl (0.07g/l) with normal IgG and IgM levels and normal antibody response in patients over 4 years of age.17
FC was defined as a patient diagnosed or followed-up by our unit in whom family screening showed there to be one member of the same family affected. If only the index case was affected, it was considered as SC.
First-degree relatives with SIgAD were denominated as affected FDR (FDR-A) and relatives in whom no SIgAD was detected were classified as healthy FDR (FDR-H).
Laboratory studiesHaematological and biochemical parameters were obtained through routine lab methods. Immunoglobulins (Ig) were determined though nephelometry, IgG subclasses through nephelometry with monoclonal antibodies (MoAb) and vaccine response in patients immunised through conventional serological methods.
They were described as serum concentrations of immunoglobulins (IgM, IgG, IgA, IgE), subclasses of IgG, and decreased isohaemagglutinins and antistreptolysins when they were at least 2 standard deviations (SD) below the mean of the age-adjusted reference values established by Jollif et al.18 for the subject. ASLO values <200UI/ml and isohaemaglutinines below 1/32 were considered negative, both for IgM and for IgG. The vaccine response was considered negative in cases with prior confirmation of the applicable vaccination and certain values below the cut point of our centre. ANA values were considered pathological when they were above 1/80.
Statistical analysisFisher's exact test was used for the analysis of qualitative data, and the Student's t test for quantitative variables, with 5% significance. For the bivariate analysis, odds ratios (OR) were calculated. The statistical analysis was carried out using SPSS (SPSS Statistics for Windows, Version 17.0, SPSS Inc., Chicago, USA).
ResultsSociodemographic characteristicsThe sample included 130 participants, 42 cases and 88 FDRs (Fig. 1).

The mean age±SD of cases with SIgAD was 11 years±4.54 and 57% were female, with no differences found between FCs and SCs (p=0.17 and 0.10, respectively). The mean age of FDRs was 34±15.85 years and 57% (50) were female. No case of consanguinity was collected.
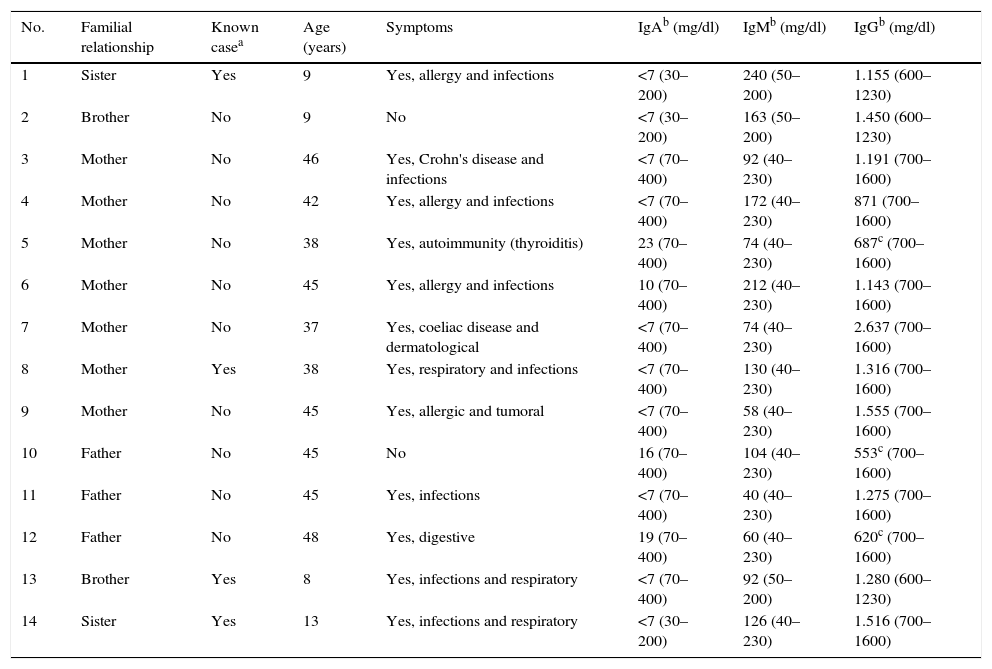
Familial casesIn 13 of the 42 patients with SIgAD (31%), 1 or more affected relatives were found. The frequency of SIgAD in FDRs was estimated as 1 case for each 6 family members. One mother, 1 brother and 2 sisters had a known previous diagnosis of SIgAD (Table 1).
Sociodemographic clinical and laboratory characteristics of ill first-degree relatives (FDR-A).
| No. | Familial relationship | Known casea | Age (years) | Symptoms | IgAb (mg/dl) | IgMb (mg/dl) | IgGb (mg/dl) |
|---|---|---|---|---|---|---|---|
| 1 | Sister | Yes | 9 | Yes, allergy and infections | <7 (30–200) | 240 (50–200) | 1.155 (600–1230) |
| 2 | Brother | No | 9 | No | <7 (30–200) | 163 (50–200) | 1.450 (600–1230) |
| 3 | Mother | No | 46 | Yes, Crohn's disease and infections | <7 (70–400) | 92 (40–230) | 1.191 (700–1600) |
| 4 | Mother | No | 42 | Yes, allergy and infections | <7 (70–400) | 172 (40–230) | 871 (700–1600) |
| 5 | Mother | No | 38 | Yes, autoimmunity (thyroiditis) | 23 (70–400) | 74 (40–230) | 687c (700–1600) |
| 6 | Mother | No | 45 | Yes, allergy and infections | 10 (70–400) | 212 (40–230) | 1.143 (700–1600) |
| 7 | Mother | No | 37 | Yes, coeliac disease and dermatological | <7 (70–400) | 74 (40–230) | 2.637 (700–1600) |
| 8 | Mother | Yes | 38 | Yes, respiratory and infections | <7 (70–400) | 130 (40–230) | 1.316 (700–1600) |
| 9 | Mother | No | 45 | Yes, allergic and tumoral | <7 (70–400) | 58 (40–230) | 1.555 (700–1600) |
| 10 | Father | No | 45 | No | 16 (70–400) | 104 (40–230) | 553c (700–1600) |
| 11 | Father | No | 45 | Yes, infections | <7 (70–400) | 40 (40–230) | 1.275 (700–1600) |
| 12 | Father | No | 48 | Yes, digestive | 19 (70–400) | 60 (40–230) | 620c (700–1600) |
| 13 | Brother | Yes | 8 | Yes, infections and respiratory | <7 (70–400) | 92 (50–200) | 1.280 (600–1230) |
| 14 | Sister | Yes | 13 | Yes, infections and respiratory | <7 (30–200) | 126 (40–230) | 1.516 (700–1600) |
Of the familial cases diagnosed, 50% (7) were mothers, 21.4% (3) fathers, 14.3% (2) brothers and 14.3% (2) sisters (p=0.55; OR=1.96; 95% CI, 0.62–6.21). In other words, 22% mothers, 14.5% brothers and sisters and 10% fathers showed SIgAD. The sociodemographic, clinical and laboratory characteristics of each FDR-A are shown separately in Table 1. Taking an estimated prevalence of SIgAD of 1/600 in Caucasians as a representation of the sample, calculations showed that 6 relatives needed to be studied to find 1 FC with SIgAD.
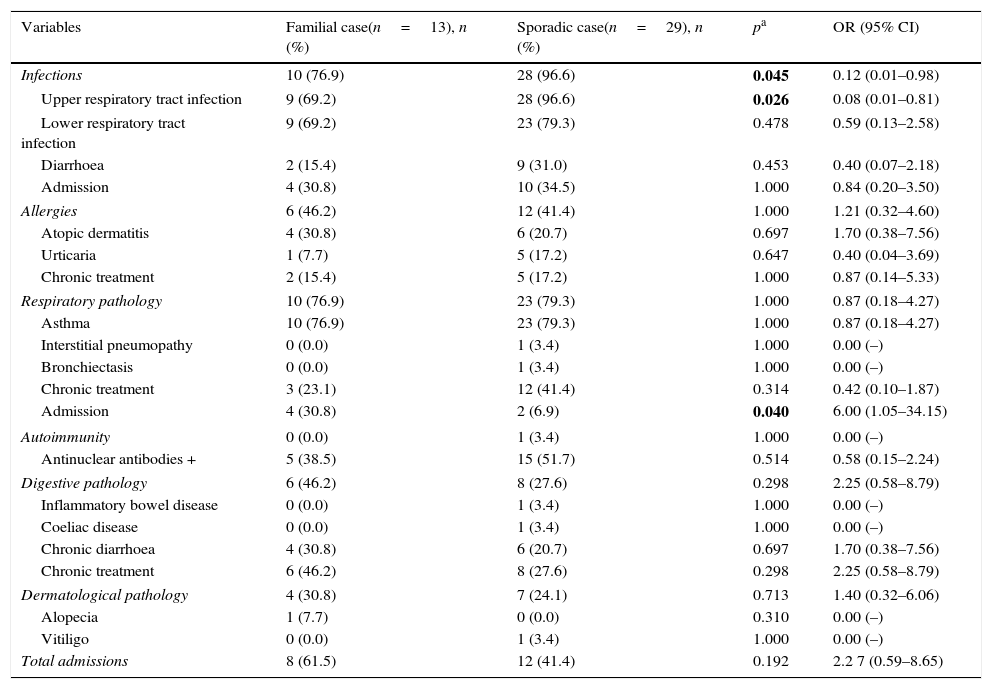
Clinical manifestationsAll children with SIgAD presented some type of clinical manifestation. The recorded infections are shown in Table 2. Parasites were isolated in only 2 (6.9%) SCs with diarrhoea.
Clinical characteristics of FCs and SCs.
| Variables | Familial case(n=13), n (%) | Sporadic case(n=29), n (%) | pa | OR (95% CI) |
|---|---|---|---|---|
| Infections | 10 (76.9) | 28 (96.6) | 0.045 | 0.12 (0.01–0.98) |
| Upper respiratory tract infection | 9 (69.2) | 28 (96.6) | 0.026 | 0.08 (0.01–0.81) |
| Lower respiratory tract infection | 9 (69.2) | 23 (79.3) | 0.478 | 0.59 (0.13–2.58) |
| Diarrhoea | 2 (15.4) | 9 (31.0) | 0.453 | 0.40 (0.07–2.18) |
| Admission | 4 (30.8) | 10 (34.5) | 1.000 | 0.84 (0.20–3.50) |
| Allergies | 6 (46.2) | 12 (41.4) | 1.000 | 1.21 (0.32–4.60) |
| Atopic dermatitis | 4 (30.8) | 6 (20.7) | 0.697 | 1.70 (0.38–7.56) |
| Urticaria | 1 (7.7) | 5 (17.2) | 0.647 | 0.40 (0.04–3.69) |
| Chronic treatment | 2 (15.4) | 5 (17.2) | 1.000 | 0.87 (0.14–5.33) |
| Respiratory pathology | 10 (76.9) | 23 (79.3) | 1.000 | 0.87 (0.18–4.27) |
| Asthma | 10 (76.9) | 23 (79.3) | 1.000 | 0.87 (0.18–4.27) |
| Interstitial pneumopathy | 0 (0.0) | 1 (3.4) | 1.000 | 0.00 (–) |
| Bronchiectasis | 0 (0.0) | 1 (3.4) | 1.000 | 0.00 (–) |
| Chronic treatment | 3 (23.1) | 12 (41.4) | 0.314 | 0.42 (0.10–1.87) |
| Admission | 4 (30.8) | 2 (6.9) | 0.040 | 6.00 (1.05–34.15) |
| Autoimmunity | 0 (0.0) | 1 (3.4) | 1.000 | 0.00 (–) |
| Antinuclear antibodies + | 5 (38.5) | 15 (51.7) | 0.514 | 0.58 (0.15–2.24) |
| Digestive pathology | 6 (46.2) | 8 (27.6) | 0.298 | 2.25 (0.58–8.79) |
| Inflammatory bowel disease | 0 (0.0) | 1 (3.4) | 1.000 | 0.00 (–) |
| Coeliac disease | 0 (0.0) | 1 (3.4) | 1.000 | 0.00 (–) |
| Chronic diarrhoea | 4 (30.8) | 6 (20.7) | 0.697 | 1.70 (0.38–7.56) |
| Chronic treatment | 6 (46.2) | 8 (27.6) | 0.298 | 2.25 (0.58–8.79) |
| Dermatological pathology | 4 (30.8) | 7 (24.1) | 0.713 | 1.40 (0.32–6.06) |
| Alopecia | 1 (7.7) | 0 (0.0) | 0.310 | 0.00 (–) |
| Vitiligo | 0 (0.0) | 1 (3.4) | 1.000 | 0.00 (–) |
| Total admissions | 8 (61.5) | 12 (41.4) | 0.192 | 2.2 7 (0.59–8.65) |
SC: sporadical cases; FC: familial cases; CI: confidence interval; OR: odds ratio.
Bold values are statistically significant.
As regards autoimmunity phenomena, only 1 case of Hashimoto thyroiditis was described. There were no neoplasias.
No significant clinical differences were found between FCs and SCs. In fact, even infections were more frequent in SCs (p=0.045, OR=0.12; 95% CI, 0.01–0.98). In contrast, FCs presented more admissions for respiratory causes than SCs (p=0.04, OR=6, 95% CI, 1.05–34.15), which is not confirmed when assessing the total number of admissions (Table 2).
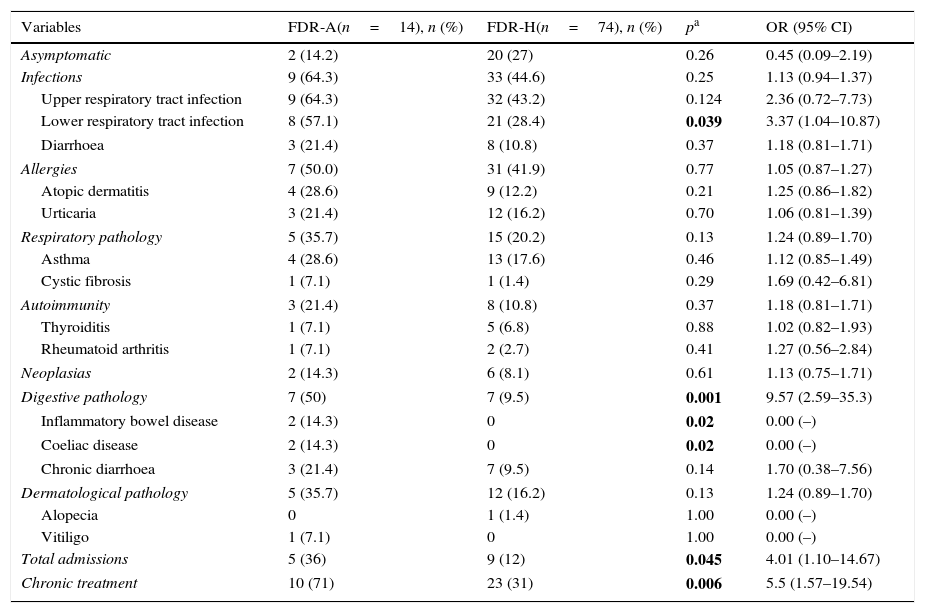
When FDRs were assessed, no differences were found as regards clinical manifestations between both groups, except for the presence of digestive pathologies, significantly more frequent in FDR-As (Table 3). Ten FDRs had psoriasis (13.5%), all within the FDR-H group. All neoplasias reported were solid tumours with no differences between both groups.
Clinical characteristics of FDR-Hs and FDR-As.
| Variables | FDR-A(n=14), n (%) | FDR-H(n=74), n (%) | pa | OR (95% CI) |
|---|---|---|---|---|
| Asymptomatic | 2 (14.2) | 20 (27) | 0.26 | 0.45 (0.09–2.19) |
| Infections | 9 (64.3) | 33 (44.6) | 0.25 | 1.13 (0.94–1.37) |
| Upper respiratory tract infection | 9 (64.3) | 32 (43.2) | 0.124 | 2.36 (0.72–7.73) |
| Lower respiratory tract infection | 8 (57.1) | 21 (28.4) | 0.039 | 3.37 (1.04–10.87) |
| Diarrhoea | 3 (21.4) | 8 (10.8) | 0.37 | 1.18 (0.81–1.71) |
| Allergies | 7 (50.0) | 31 (41.9) | 0.77 | 1.05 (0.87–1.27) |
| Atopic dermatitis | 4 (28.6) | 9 (12.2) | 0.21 | 1.25 (0.86–1.82) |
| Urticaria | 3 (21.4) | 12 (16.2) | 0.70 | 1.06 (0.81–1.39) |
| Respiratory pathology | 5 (35.7) | 15 (20.2) | 0.13 | 1.24 (0.89–1.70) |
| Asthma | 4 (28.6) | 13 (17.6) | 0.46 | 1.12 (0.85–1.49) |
| Cystic fibrosis | 1 (7.1) | 1 (1.4) | 0.29 | 1.69 (0.42–6.81) |
| Autoimmunity | 3 (21.4) | 8 (10.8) | 0.37 | 1.18 (0.81–1.71) |
| Thyroiditis | 1 (7.1) | 5 (6.8) | 0.88 | 1.02 (0.82–1.93) |
| Rheumatoid arthritis | 1 (7.1) | 2 (2.7) | 0.41 | 1.27 (0.56–2.84) |
| Neoplasias | 2 (14.3) | 6 (8.1) | 0.61 | 1.13 (0.75–1.71) |
| Digestive pathology | 7 (50) | 7 (9.5) | 0.001 | 9.57 (2.59–35.3) |
| Inflammatory bowel disease | 2 (14.3) | 0 | 0.02 | 0.00 (–) |
| Coeliac disease | 2 (14.3) | 0 | 0.02 | 0.00 (–) |
| Chronic diarrhoea | 3 (21.4) | 7 (9.5) | 0.14 | 1.70 (0.38–7.56) |
| Dermatological pathology | 5 (35.7) | 12 (16.2) | 0.13 | 1.24 (0.89–1.70) |
| Alopecia | 0 | 1 (1.4) | 1.00 | 0.00 (–) |
| Vitiligo | 1 (7.1) | 0 | 1.00 | 0.00 (–) |
| Total admissions | 5 (36) | 9 (12) | 0.045 | 4.01 (1.10–14.67) |
| Chronic treatment | 10 (71) | 23 (31) | 0.006 | 5.5 (1.57–19.54) |
FDR-A: affected first-degree relatives; FDR-H: healthy first-degree relatives; CI: confidence interval; OR: odds ratio.
Bold values are statistically significant.
As a marker for clinical severity, the need for hospital admission and chronic treatment was estimated. A significantly higher proportion of total admissions in FDR-As was shown with respect to FDR-Hs, with an increased overall risk of admission 4 times higher. The reasons for admission in FDR-As were infectious causes (2), respiratory (2) and 1 case secondary to digestive pathology. In FDR-Hs, 8 of the 9 admissions were caused by infections and the remainder by digestive pathology. Furthermore, 5 times more FDR-As needed chronic than FDR-Hs (Table 3). In FDR-As, most (7) were due to digestive pathology and the rest received chronic treatment for respiratory (2) and allergic (1) pathology. The usual treatment reported in FDR-Hs was for gastrointestinal alterations in 12 cases (52%), for respiratory pathology in 7 (31%) cases, and for allergies in 4 (17%) cases.
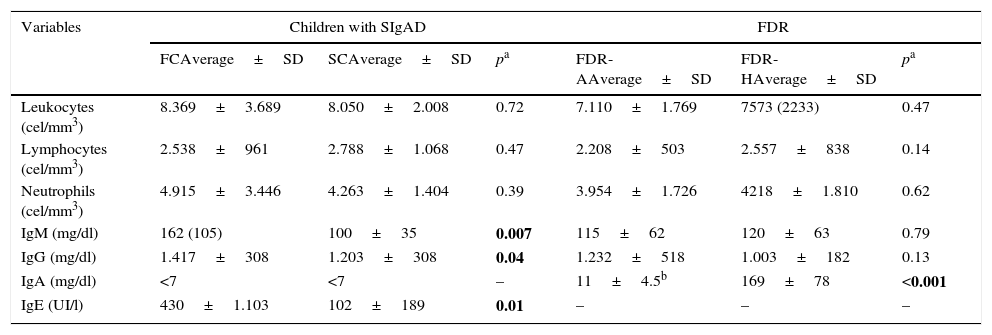
Laboratory studiesThe levels of immunoglobulins, total leukocytes, lymphocytes and neutrophils of cases and FDRs are shown in Table 4. IgG, IgM and IgE were significantly higher in FCs than SCs (p=0.007, p=0.04 and p=0.01, respectively). One patient from each group presented low levels of IgG subclasses (both IgG2). The humoral response was very similar in both groups and is shown in Table 5.
Complete blood count and serum concentrations of immunoglobulins in FC vs. SC and FDR-H vs. FDR-A.
| Variables | Children with SIgAD | FDR | ||||
|---|---|---|---|---|---|---|
| FCAverage±SD | SCAverage±SD | pa | FDR-AAverage±SD | FDR-HAverage±SD | pa | |
| Leukocytes (cel/mm3) | 8.369±3.689 | 8.050±2.008 | 0.72 | 7.110±1.769 | 7573 (2233) | 0.47 |
| Lymphocytes (cel/mm3) | 2.538±961 | 2.788±1.068 | 0.47 | 2.208±503 | 2.557±838 | 0.14 |
| Neutrophils (cel/mm3) | 4.915±3.446 | 4.263±1.404 | 0.39 | 3.954±1.726 | 4218±1.810 | 0.62 |
| IgM (mg/dl) | 162 (105) | 100±35 | 0.007 | 115±62 | 120±63 | 0.79 |
| IgG (mg/dl) | 1.417±308 | 1.203±308 | 0.04 | 1.232±518 | 1.003±182 | 0.13 |
| IgA (mg/dl) | <7 | <7 | – | 11±4.5b | 169±78 | <0.001 |
| IgE (UI/l) | 430±1.103 | 102±189 | 0.01 | – | – | – |
SC: sporadical cases; FC: familial cases; SD: standard deviation; SIgAD: selective IgA deficiency; FDR: first-degree relatives; FDR-A: affected FDR; FDR-H: Healthy FDR.
Bold values are statistically significant.
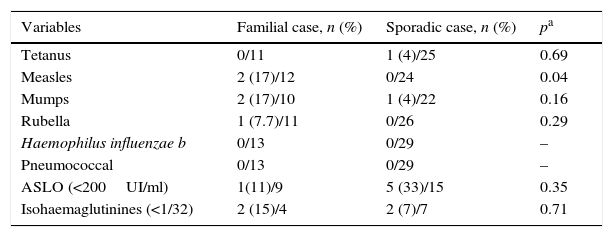
Study of the production of antibodies in familial and sporadic cases.
| Variables | Familial case, n (%) | Sporadic case, n (%) | pa |
|---|---|---|---|
| Tetanus | 0/11 | 1 (4)/25 | 0.69 |
| Measles | 2 (17)/12 | 0/24 | 0.04 |
| Mumps | 2 (17)/10 | 1 (4)/22 | 0.16 |
| Rubella | 1 (7.7)/11 | 0/26 | 0.29 |
| Haemophilus influenzae b | 0/13 | 0/29 | – |
| Pneumococcal | 0/13 | 0/29 | – |
| ASLO (<200UI/ml) | 1(11)/9 | 5 (33)/15 | 0.35 |
| Isohaemaglutinines (<1/32) | 2 (15)/4 | 2 (7)/7 | 0.71 |
Despite the fact that there are no differences in IgG and IgM levels between both groups of FDRs, 3 cases of hypogammaglobulinaemia G were detected in FDR-As and none in FDR-H cases (Table 4).
DiscussionWe found no evidence of the presence of more severe clinical phenotypes in familial cases of SIgAD in our series. We were only able to prove a clearly higher risk of admission due to respiratory pathology (most for asthma) in FCs, which could indicate a more severe phenotype; however, this was not confirmed when making an overall assessment of admissions for any cause in both groups.
In any event, a number of factors must be taken into consideration. Although patients with SIgAD are asymptomatic in most of the series reported in the literature,2,19 in our study all patients reported some kind of clinical sign, possibly due to selection bias in the centre where they received treatment. This may have contributed to the absence of significant clinical differences in our sample. On the other hand, loss of information is always a risk in retrospective studies, especially as regards minor clinical manifestations such as mild infections.
The distribution of infection by type did not differ to that reported in the literature5,16 and, despite the fact that overall infections were more frequent in SCs, there were no differences in the need for hospital admission, which implies a mild course in both groups. The low prevalence of allergic processes and intestinal, autoimmune and tumoral pathology in our sample compared with other series1,2,15,16,19–24 — probably related to the fact that it is a paediatric population — prevents us from reaching conclusions as regards the role of familial aggregation as a risk factor for developing this type of pathology. However, the high prevalence of positive ANA in patients in both groups with no evident autoimmune manifestations was striking and could be related to SIgAD as an epiphenomenon of autoimmune disorders that can increase the risk of developing an autoimmune disorder in adulthood, or as part of a compensatory response to infection and to a sustained condition, as shown by Fusaro et al.25 Likewise, total IgE levels were also higher in FCs despite not presenting a higher prevalence of clinically significant allergic processes.
Serum concentrations of IgG and IgM were significantly higher in FCs. There was no obvious reason for this, as prevalence of severity of infection or inflammation was not higher in this group.
Humoral response, measured through isohaemagglutinins and titres of ASLO, was poor in a significant number of patients (3 FCs and 6 SCs), so it would be convenient to make these determinations in all cases. The response to vaccines included in our vaccination schedule (tetanus, measles, mumps, rubella, H. influenzae and S. pneumoniae) was good in both groups, unlike the measles vaccine, where bad response was only reported in FCs. Irrespective of their group, we consider that these patients must be closely followed-up to assess whether these findings can predict the development of more clinically significant immunodeficiencies such as CVID, as reported in other publications.11
As regards the usefulness of family screening, it seems reasonable to affirm that patients detected based on the familial screening of paediatric patients with SIgAD present sufficiently significant clinical symptomatology to justify this strategy, particularly considering the high prevalence of familial cases shown in this and other studies.3,26,27
Thus, the most significant risk factor for the development of SIgAD shown in previous studies is the existence of a positive family history.28 Due to ethnicity-related genetic difference, the prevalence of affected family members no doubt differs in different regions of the world.3 In our analysis, the frequency of one or more FDR affected was 16%, diagnosed de novo in more than 70% of relatives. There was more than 1 case in the same family in 31% of cases. This percentage is similar to other series, which estimate figures of up to 32.4%, with an elevated prevalence of consanguinity not found in our sample3 and considerably higher than the general population, with a prevalence among Caucasians of 1 case in every 600.
Our study shows that the risk of SIgAD in FDRs of patients with this pathology was slightly higher in mothers (22%) than fathers (10%) and siblings (14.5%), although no major variations were found. These data are not consistent with previous studies, which report a higher risk in siblings.3,27,28 Vorechovský et al.28 analysed serum concentrations of Ig in close relatives of Swedish patients with SIgAD and CVID, estimating the relative risk for the siblings of patients with SIgAD to be approximately 50. However, parent of origin penetrance differences have been found in the transmission of SIgAD to children.29 Some investigations have found a higher prevalence of children with SIgAD born from mothers with this pathology than from fathers with the same defect.28,30 This type of inheritance with more predominance in maternal transmission could be related to mitochondrial defects and could explain in part the higher percentage of affected mothers found in our study, since the number of siblings (one brother/sister for each mother) is small in relation to other studies.3,27
Despite the fact that in our study only 14% of FDR-As were asymptomatic, this did not differ significantly from FDR-Hs, which is why it could be assumed that pathologies reported in FDR-As were similar to those in the general population. In 2008, Aghamohammadi et al.27 presented a study which affirmed that all relatives of patients with SIgAD who also had this defect were asymptomatic; however, they were not compared with the cohort of healthy relatives, which may explain our results. A recent case–control study comparing the symptoms of SIgAD patients with those of healthy patients found higher rates of infections, allergic phenomena and autoimmunity in patients with SIgAD26; we were unable to confirm this in our series. However, taking hospital admissions and need for chronic treatment as a marker of clinical severity would support the finding of Jorgensen et al.26
Although we were unable to show the significantly lower levels of IgG and IgM reported by other authors, who propose these variables as markers of risk for evolution to CVID, we detected 3 adult patients with hypogammaglobulinaemia who should be studied thoroughly.3,27 These findings, however, cannot be fully validated because IgG subclasses and antibody levels in family members were not evaluated.
In conclusion, we consider that the elevated prevalence of SIgAD in FDRs of patients with this pathology associated with a greater clinical severity, measured indirectly through admission rates and the need for chronic treatment, shows FDRs could benefit from screening programmes. Since few relatives will need to be screened to find one positive case, this could be a cost-effective prevention strategy. This would enable individuals with this type of immunodeficiency to start treatment or prophylaxis measures to prevent hospitalisation, or to simply attend regular check-ups due to the risk of evolution to CVID.
Authors’ contributionP. Soler-Palacín and E. Cobos-Carrascosa have contributed equally to the study.
Conflicts of interestThe authors declare that there are no conflicts of interest.
Please cite this article as: Soler-Palacín P, Cobos-Carrascosa E, Martín-Nalda A, Caracseghi F, Hernández M, Figueras-Nadal C. ¿Es útil el cribado familiar en el déficit selectivo de inmunoglobulina A? An Pediatr (Barc). 2016;84:70–78.
