





Analizar las características epidemiológicas, clínicas y evolutivas de la epilepsia rolándica para facilitar su sospecha diagnóstica en la práctica clínica diaria.
Pacientes y métodosSe han revisado 56 historias clínicas de pacientes con epilepsia rolándica y se han registrado características epidemiológicas y clínicas, exploraciones complementarias y datos evolutivos. Los criterios diagnósticos aplicados fueron los de la International League Against Epilepsy.
ResultadosLa edad media al diagnóstico era de 7,7 años. El 62,5 % fueron diagnosticados en edad escolar, con mayor prevalencia del sexo masculino (58,9 %). El 80,4 % de los pacientes tuvieron crisis exclusivamente durante el sueño caracterizadas por contracciones hemifaciales con desviación ocular y/o cefálica (76,8 %), sialorrea (44,6 %), sonidos guturales (30,6 %), crisis motoras secundariamente generalizadas (35,7 %) y/o hemicorporales (26,8 %), disartria (17,9 %) y parestesias unilaterales (16,1 %). Se constataron paroxismos de localización centrotemporal, preferentemente unilaterales (78,6 %). El 50,7 % de las recurrencias se dieron en los primeros 12 meses tras el diagnóstico, el 24,6 % entre los 12 y 24 meses, y el 24,6 % restante entre los 2 y 4 años. Se objetivaron 2 casos de evolución atípica: uno de actividad continua de punta-onda en sueño lento, y otro de afasia adquirida (síndrome de Landau-Kleffner).
ConclusionesLa epilepsia rolándica constituye un síndrome epiléptico específicamente pediátrico que afecta preferentemente a varones en edad escolar. Su secuencia semiológica es bastante característica, y es imprescindible documentar paroxismos centrotemporales para su diagnóstico. Su pronóstico es excelente; sin embargo, dado que algunos pacientes cursan una evolución atípica y/o una afectación cognitiva transitoria sería conveniente mantener un riguroso control evolutivo.
To analyse the epidemiological, clinical and developmental characteristics of Rolandic epilepsy as an aid to its suspected diagnosis in daily clinical practice.
Patients and methodsThe medical records of 56 patients with Rolandic epilepsy were reviewed in order to collect epidemiological and clinical features, results of complementary examinations and developmental data. The criteria defined by the International League Against Epilepsy (ILAE) were used in the diagnosis
ResultsMean age at diagnosis was 7.7 years. In all, 62.5 % were diagnosed at school age, with a higher prevalence of males (58.9 %). Seizures occurred during sleep in 84.4 % of patients, and they were mainly characterised by hemifacial seizures with eye deviation and/or headaches (76.8 %), hypersalivation (44.6 %), guttural sounds (30.6 %), secondary generalised tonic-clonic (35.7 %) and/or unilateral clonic or tonic seizures (26.8 %), dysarthria (17.9 %) and unilateral paresthesias (16.1 %). Inter-ictal EEG showed paroxysms in the centrotemporal regions, frequently unilateral (78.6 %). Of all recurrences, 50.7 % occurred during the first 12 months after diagnosis, 24.6 % between 12 and 24 months after diagnosis, and 24.6% between 2 and 4 years of follow up. Two patients with atypical progression were recorded: a case with epilepsy with continuous spikes and waves during slow-wave sleep, and another case with a Landau-Kleffner syndrome.
ConclusionsRolandic epilepsy is a common type of epilepsy in the pediatric age group and generally begins at school-aged children. Its semiological sequence is fairly characteristic, and finding centrotemporal spikes is considered as necessary for the syndromic diagnosis. The prognosis is excellent; however, as a few patients may progress to atypical outcomes and/or neuropsychological deficits, a rigorous developmental control of these patients should be of the highest priority.
Las epilepsias parciales idiopáticas suponen el 18,3–20,5 % de las epilepsias infantiles y, en consecuencia, representan una carga asistencial importante en la práctica clínica pediátrica1,2. Según el nuevo esquema diagnóstico de las epilepsias y síndromes epilépticos propuesto por la ILAE (International League Against Epilepsy), la epilepsia parcial benigna infantil con paroxismos centrotemporales o epilepsia rolándica quedaría incluida junto con la epilepsia occipital benigna de la infancia de comienzo precoz (síndrome de Panayiotopoulos) y tardío (tipo Gastaut), en el grupo de las epilepsias parciales idiopáticas infantiles3–5. El concepto de benignidad atribuido a este grupo de síndromes epilépticos hace referencia a la ausencia de déficit neurológico y/o cognitivo y normalidad neurorradiológica y, especialmente, a su tendencia a remitir de forma espontánea en la adolescencia, probablemente en relación con las modificaciones estructurales y funcionales genéticamente determinadas que el cerebro va experimentando con la edad6–10. Por tanto, su diagnóstico y/o clasificación permitiría, con un amplio margen de seguridad, vaticinar un curso evolutivo favorable11; aunque se han descrito diversos trastornos cognitivos y/o conductuales transitorios relacionados con la actividad paroxística intercrítica12–15 que han dado lugar a que algunos autores hayan sugerido una nueva variante o “epilepsia parcial benigna infantil con paroxismos centrotemporales-plus”16.
La epilepsia rolándica constituye uno de los síndromes epilépticos infantiles más comunes, particularmente en la edad escolar1,2,17–19. Las crisis generalmente son breves y tienen lugar durante el sueño, y clásicamente se describen como manifestaciones sensitivomotoras de localización hemifacial y/o fonatorias en relación con la topografía de la descarga rolándica y que, en menor medida, se acompañan de crisis motoras hemicorporales y/o generalizadas. Es imprescindible documentar paroxismos centrotemporales para confirmar el diagnóstico3–5,20–23.
El objetivo del presente trabajo consiste en exponer y analizar las características epidemiológicas, clínicas y evolutivas de un grupo de pacientes con epilepsia rolándica en orden a facilitar su sospecha diagnóstica en la práctica clínica diaria.
MATERIAL Y MÉTODOSSe han revisado de forma retrospectiva 56 historias clínicas de pacientes diagnosticados de epilepsia rolándica en la Unidad de Neuropediatría del Hospital Virgen del Camino de Pamplona –siempre y cuando el tiempo de seguimiento evolutivo fuera superior a los 2 años-, y que acudieron a su control evolutivo ambulatorio entre enero y diciembre de 2006. Para su diagnóstico y clasificación se han aplicado los criterios actualizados de la ILAE3,4.
De cada historia clínica se registraron datos epidemiológicos: sexo, edad en el momento del diagnóstico, antecedentes personales (embarazo y parto, período neonatal, desarrollo psicomotor y convulsiones febriles) y familiares (convulsiones febriles y epilepsia); datos clínicos: horario, frecuencia y semiología de las crisis, junto con exámenes complementarios: electroencefalograma (EEG) en reposo y tras activaciones (hiperventilación y estimulación luminosa intermitente) y estudios de neuroimagen -tomografía computarizada (TC) y/o resonancia magnética (RM) craneal-, y datos evolutivos: fármacos antiepilépticos empleados, respuesta clínica y del EEG al tratamiento, supresión farmacológica, recaídas y tiempo de seguimiento.
Tras el diagnóstico, la totalidad de pacientes incluidos en la muestra fueron citados al mes y, posteriormente, salvo cambios evolutivos significativos, a los 3, 6 y 12 meses, y luego cada 6 meses.
Los resultados se expresan como medias y porcentajes con sus desviaciones estándar y/o intervalos de confianza. Para el análisis estadístico (t de Student, comparación de proporciones) se utilizó el programa informático SPSS 12.0 para Windows.
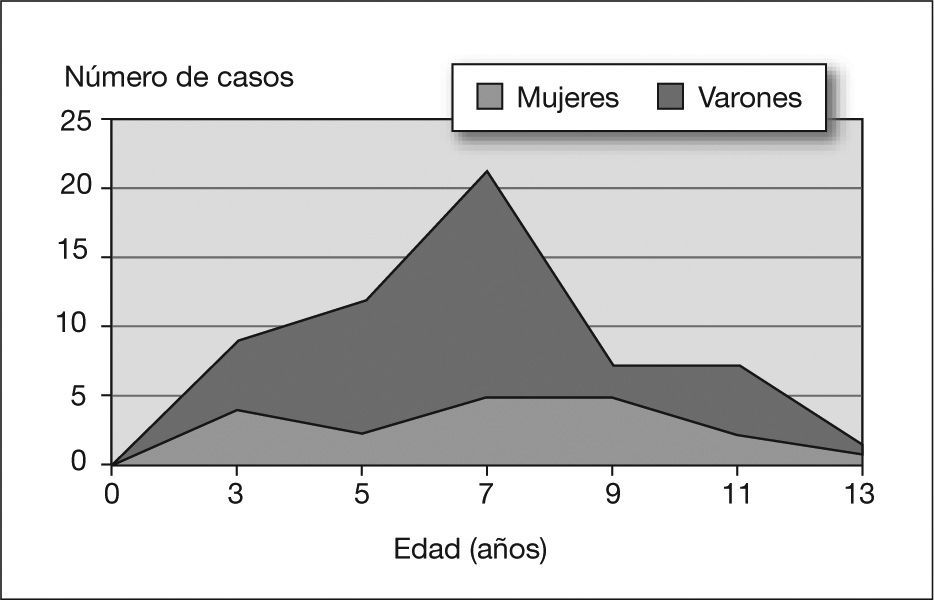
RESULTADOSCaracterísticas epidemiológicasEn la tabla 1 se exponen parte de los datos epidemiológicos registrados de los 56 pacientes incluidos en la muestra. La relación varón/mujer era de 1,5. Las edades correspondientes al diagnóstico oscilaban entre 2,9 y 13,8 años (mediana: 8,4 años), y seguían una curva de distribución normal con un pico máximo en el intervalo de edad de 7–8 años (fig. 1). El 62,5 % de los casos (n = 35) fueron diagnosticados en edad escolar, mientras que en edad preescolar y adolescencia lo fueron el 25% (n = 14) y el 12,5 % (n = 7), respectivamente. La historia familiar de epilepsia era positiva en 15 casos (26,8 %), y en 9 de ellos se trataba de epilepsias infantiles.
Datos epidemiológicos
| Ítems | n (%) |
| Sexo | |
| Mujeres | 23 (41,1) |
| Varones | 33 (58,9) |
| Convulsiones febriles | |
| Personales | 6 (10,7) |
| Familiares | 5 (8,9) |
| Total | 11 (19,6) |
| Epilepsia familiar | |
| 1 .er grado | 1 (1,8) |
| 2.o grado | 14 (25,0) |
| Total | 15 (26,8) |
| Ítems | Media (IC 95%) |
| Edad (años) | |
| En el momento del diagnóstico | 7,7 (7,0-8,4) |
| En la última crisis | 9,2 (8,5-9,9) |
| En el último control | 13,3 (12,4-14,2) |
| Tiempo de seguimiento | 5,6 (4,8-6,4) |
IC 95 %: intervalo de confianza del 95 %.
La totalidad de los pacientes procedían de embarazos a término, salvo un caso de prematuridad (33 semanas de gestación y peso de 1.830g). El 83,9 % de los partos (n = 47) fueron eutócicos y el 16,1 % restantes terminaron instrumentados (2 casos) o por cesárea (7 casos). El peso al nacer oscilaba entre 2.250 y 4.800g, exceptuando al recién nacido pretérmino. El período neonatal fue normal en la totalidad de casos, salvo en dos pacientes: uno de ellos presentó convulsiones neonatales, y otro, el recién nacido pretérmino, presentó una enterocolitis necrotizante que precisó resección quirúrgica. El desarrollo psicomotor fue normal en la totalidad de los casos; no obstante, con anterioridad al diagnóstico, dos pacientes presentaban dificultad de aprendizaje: uno de ellos asociado a una hipoacusia neurosensorial grave bilateral y el otro con un retraso escolar de causa inespecífica.
Manifestaciones clínicasEl número medio de crisis por paciente fue de 3,8 (intervalo de confianza [IC 95 %]: 2,9-4,7). Un total de 14 pacientes (25 %) presentaron solamente una crisis, 29 (51,8 %) entre dos y cinco crisis y los 13 restantes (23,2 %) un número superior, aunque tan sólo 4 tuvieron más de 10. Un total de 45 pacientes (80,4 %) presentaron crisis exclusivamente durante el sueño (30 casos) o al despertar (15 casos), 9 (16,1 %) en vigilia y 2 (3,6 %) de manera indistinta; 36 pacientes (64,2 %) presentaron exclusivamente crisis parciales simples (25 casos) o complejas (11 casos), y los 20 restantes (35,7 %) crisis secundariamente generalizadas. A lo largo del curso evolutivo de los 56 pacientes incluidos en el estudio quedaron registradas 206 crisis. Entre las características clínicas, y en orden de frecuencia, destacaban las siguientes: contracciones tónicas y/o clónicas hemifaciales con desviación de la comisura labial (76,8 %), sialorrea (44,6 %), ruidos guturales (39,3 %), crisis tónico-clónicas generalizadas (35,7 %), hemiclonías de extremidad superior y/o inferior (26,8 %), disartria o anartria (17,9 %), parestesias hemifaciales y/o hemicorporales (16,1 %) y automatismos orofaciales (7,1 %). La duración media de las crisis fue de 2,8min, y el 73,2% no llegaban a los 5min. En dos pacientes (3,6 %) la duración de su primera crisis fue superior a los 30min (estado convulsivo). Hubo dos pacientes que consultaron por presentar salvas de parpadeo sin pérdida de conciencia (5–6 diarias) y desviación oculocefálica esporádica, de 2–3 semanas de evolución; y otro, por haber presentado dos episodios de pérdida de conciencia, de corta duración, sin movimientos tónico-clónicos. En un paciente se constató una hemiparesia poscrítica, que ya no se observó en posteriores recurrencias, y que fue recuperándose paulatinamente en el transcurso de 1h.
Hallazgos electroencefalográficosEn todos los pacientes el trazado interictal mostraba una actividad de fondo sincronizada con una focalidad epileptiforme rolándica. Se trataba de una actividad paroxística, bien en forma de complejos “punta-onda” (42 casos) y, en menor proporción, como “polipunta-onda” (14 casos), de localización centrotemporal unilateral (78,6 %) -izquierda (23 casos) y derecha (21 casos)– o bilateral (21,4 %). En 5 casos (8,9 %) se evidenciaba una tendencia a la generalización y en 8 casos (14,3 %) una difusión a regiones contralaterales homónimas (4 casos) o adyacentes (4 casos). En 21 pacientes (37,5 %) el EEG también fue realizado en condiciones de somnolencia o sueño, objetivándose una intensificación de la actividad paroxística (fig. 2), e incluso fue la manera de evidenciar actividad rolándica en dos casos con EEG basales normales. La hiperventilación y fotoestimulación fueron siempre negativas. A lo largo del curso evolutivo se objetivaron migraciones contralaterales del foco rolándico (6 casos), así como paroxismos multifocales simultáneos de diversa localización: frontal izquierda (2 casos), frontotemporal izquierda (2 casos), temporal izquierda (2 casos), temporoparietal derecha (1 caso) y occipital bilateral (1 caso).
Estudios de neuroimagen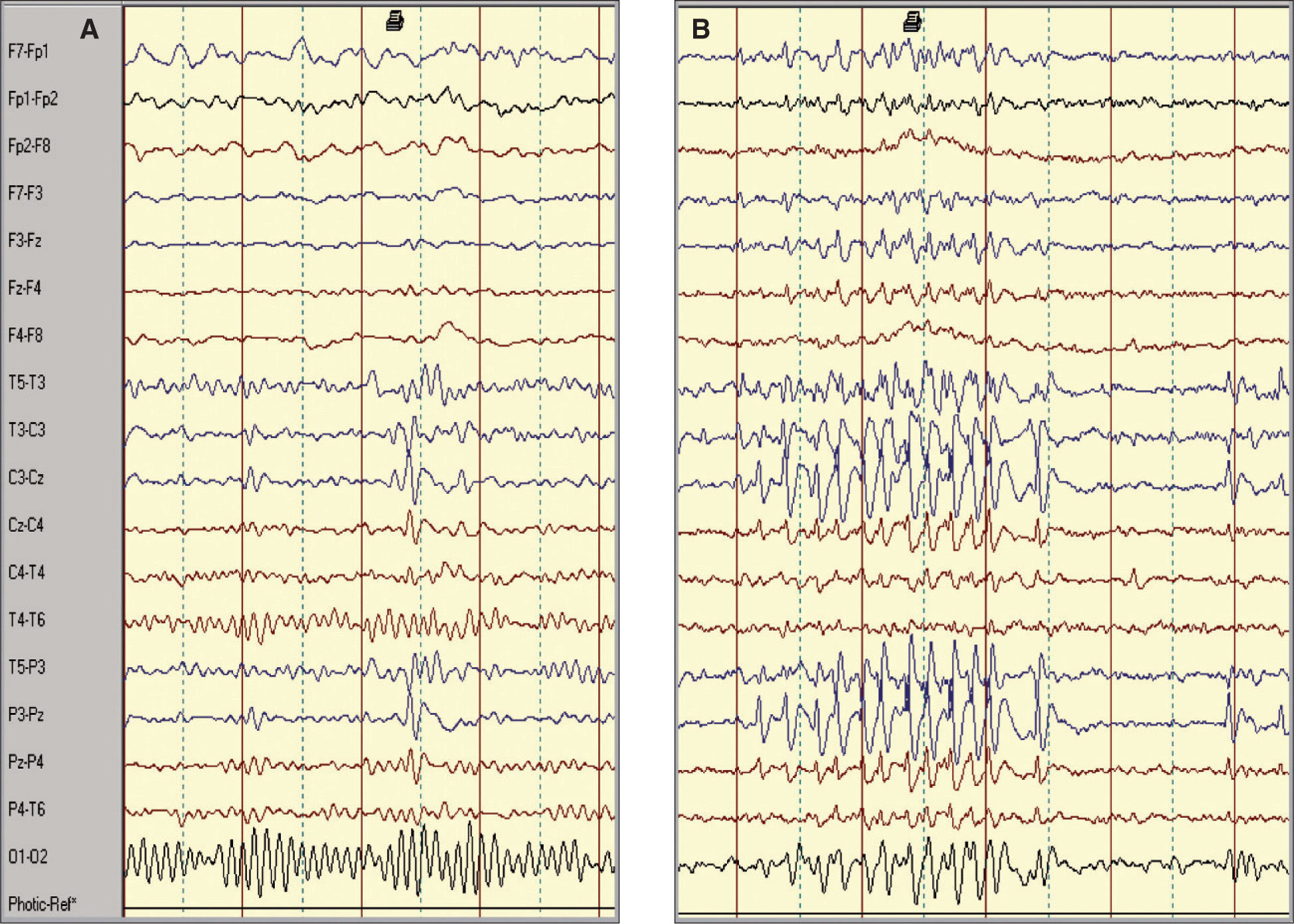
Se realizaron estudios de neuroimagen a 52 pacientes (92,9 %): TC en 37 casos (66,1 %), RM en 35 casos (62,5%) y ambas en 20 casos (35,7 %). No se encontraron hallazgos significativos, salvo en 2 casos, que presentaron, respectivamente, un angioma venoso en el lóbulo temporal derecho y un quiste aracnoideo en fosa posterior.
TratamientoEn 27 pacientes (48,2 %) se prescribieron fármacos antiepilépticos. Inicialmente se utilizaron valproato (13 casos), clobazam (5 casos), carbamacepina (4 casos), fenobarbital (3 casos) y oxcarbacepina (2 casos). Posteriormente, se realizaron diversos cambios: asociación al valproato de clobazam (2 casos), y sustituciones de valproato por clobazam (2 casos), clobazam por lamotrigina (1 caso) y carbamacepina por valproato (2 casos). El tratamiento fue retirado en 15 casos, tras una duración media de 3,9 años (IC 95 %: 3,0-4,8). Tras su retirada y seguimiento medio de 2,8 años (IC 95 %: 2,0-3,6) no hubo ninguna recidiva.
PronósticoEl 50,7 % de las recurrencias se dieron en los primeros 12 meses tras el diagnóstico, el 24,6 % entre los 12 y 24 meses, y el 24,6 % restante entre los 2 y 4 años, a excepción del caso de una paciente que tuvo una crisis a los 7,2 años del diagnóstico (a los 17,4 años de edad). Respecto a los hallazgos electroencefalográficos, al año de evolución la actividad paroxística se había normalizado en 22 casos (39,3 %) y en 15 casos (26,8 %) se normalizó más allá de los 3 años de evolución. En 7 casos (12,5 %) tras la normalización del EEG se constató una actividad paroxística rolándica en controles posteriores.
Entre los 54 pacientes con rendimiento escolar normal en el momento del diagnóstico se dieron 3 casos de trastorno de aprendizaje a lo largo del curso evolutivo: en uno ellos se normalizó al suspender el tratamiento (valproato); en otro se retiró el valproato, pero hubo que iniciar tratamiento con metilfenidato con mejoría significativa de su rendimiento escolar y, por último, un tercero presentó un trastorno conductual y/o de déficit de atención transitorio que se resolvió con apoyo escolar.
Evoluciones atípicasCabe resaltar el curso evolutivo atípico en 2 casos, cuyos antecedentes perinatales y neurológicos, así como los estudios de neuroimagen, habían sido normales:
- 1.
Una niña diagnosticada a los 2,9 años de edad y en tratamiento con valproato que 14 meses más tarde (a los 4,3 años de edad), comenzó con dificultades escolares, múltiples crisis nocturnas motoras unilaterales y/o ausencias con automatismos orofaciales. También mostraba un EEG en vigilia con paroxismos centro-temporales bilaterales y, en sueño lento, con punta-onda continua y difusa, que respondió favorablemente a la politerapia (prednisona, lamotrigina, topiramato y clobazam), pero que dejó como secuela un deterioro cognitivo que precisa apoyo psicopedagógico y adaptación curricular (edad actual: 9,1 años).
- 2.
Un varón diagnosticado a los 8,5 años de edad y en tratamiento con valproato que 9 meses más tarde (a los 9,3 años de edad) comenzó con deterioro progresivo del lenguaje y afasia, con un EEG con actividad paroxística generalizada y foco lento temporal izquierdo, que respondió favorablemente al tratamiento (clobazam) con normalización del EEG y recuperación completa del lenguaje y de su actividad escolar (edad actual: 13,6 años).
Las epilepsias benignas de la infancia constituyen síndromes epilépticos focales e idiopáticos. Se trata de cuadros genéticamente determinados que se manifiestan en pacientes con normalidad neuropsicológica como crisis parciales y/o secundariamente generalizadas, con alteraciones electroencefalográficas focales, sin lesiones neuroanatómicas evidentes y con tendencia a la remisión espontánea4,10,20,22–24. No obstante, se han descrito otras modalidades de epilepsia parcial idiopática y/o benigna de la infancia, tales como la epilepsia parcial benigna con sintomatología afectiva, epilepsia parcial benigna con paroxismos inducidos en áreas parietales, epilepsia parcial idiopática con potenciales evocados somatosensoriales gigantes, epilepsia parcial benigna con paroxismos frontales y epilepsia parcial benigna atípica25–27. Sin embargo, la nueva Clasificación de Epilepsias y Síndromes Epilépticos3–5 tan sólo reconoce como variantes bien diferenciadas de epilepsia benigna de la infancia a la epilepsia rolándica y la epilepsia con paroxismos occipitales de inicio precoz (síndrome de Panayiotopoulos) y de inicio tardío (tipo Gastaut).
Entre los aspectos epidemiológicos de esta serie cabe destacar que la edad correspondiente al momento del diagnóstico oscilaba entre 2,9 y 13,8 años, y seguía una distribución normal con un pico de máxima incidencia en la edad escolar y con un sensible predominio del sexo masculino (cociente varón/mujer: 1,5), lo que coincide con los datos publicados4,10,11,20,28,29. Cabe señalar que el 16,1 % de los casos fueron diagnosticados en edades relativamente tempranas, entre los 2,9 y 4,8 años de edad, sin que por ello tuvieran distinta evolución y/o pronóstico, lo que contrasta con los datos aportados por algunos autores relacionando su inicio precoz con mayor riesgo de presentar múltiples recurrencias30,31.
En el 26,8 % de los pacientes de esta serie figuraban antecedentes familiares de epilepsia y, generalmente, se trataba de familiares de segundo grado -a excepción del caso de la madre de un paciente– que habían presentado epilepsia exclusivamente en la edad infantil. Aunque el carácter retrospectivo condiciona la rigurosidad del registro anamnésico, cabe pensar que estos familiares, en gran parte, habrían presentado en su infancia epilepsias rolándicas o de naturaleza benigna. La alta prevalencia de antecedentes familiares con trastornos paroxísticos que suelen observarse en estos pacientes sugiere una predisposición genética; no obstante, la identificación de distintas mutaciones génicas en estos pacientes suscita que se trataría de un mecanismo genético complejo y/o heterogéneo32–35.
La secuencia semiológica que permite sospechar y/o reconocer este síndrome, a pesar de haberse descrito distintas variantes semiológicas según la topografía anatómica de la descarga rolándica, comprende una serie de manifestaciones bastante específicas, tales como signos motores orofaciales (contracciones tónicas y/o clónicas hemifaciales), bloqueo del habla (disartria), sialorrea y sintomatología somatosensitiva (parestesias unilaterales), que han sido consideradas criterios diagnósticos4,10,11,20–24. En esta serie, la mayoría de los pacientes presentaron las crisis durante el sueño y/o al despertar, tal como se refiere en la literatura médica10,20,28–30, y se caracterizaron preferentemente por desviación tónica de la comisura bucal hacia un lado y clonias hemifaciales que, en menor proporción, se acompañaban de manifestaciones fonatorias (sonidos guturales, imposibilidad para articular palabras, etc.). La sialorrea es un signo muy común en estos pacientes, aunque no queda claro si se trataría de un fenómeno autonómico o más bien secundario a la afectación ictal de la musculatura faríngea y/o laríngea y, en consecuencia, a una dificultad para su deglución10. Aunque la sintomatología somatosensitiva (parestesias unilaterales que afectan a la lengua, encías, labios, extremidades superiores y/o inferiores) se describe como un signo ictal que suele preceder, e incluso despertando al paciente, a los signos motores, en esta serie tan sólo han quedado registradas en el 16,1 % de las crisis, posiblemente debido al carácter retrospectivo de la recogida de datos. Sin embargo, el registro porcentual de crisis que evolucionaron a clonias unilaterales de extremidades superiores (crisis faciobraquial) y/o inferiores o se generalizaron (rigidez y/o movimientos tónico-clónicos) fue similar al descrito por otros autores4,10,24. En un alto porcentaje de casos, el 44,6 % de los pacientes en esta serie, las crisis eran parciales simples; es decir, con sintomatología sensitiva-motora-fonatoria sin afectación del nivel de conciencia, lo que les permitía describir lo ocurrido. En el resto de casos, bien por tratarse de crisis parciales complejas o por una generalización de las crisis presumiblemente temprana, los pacientes no recordaban lo sucedido. El síncope ictal, si bien suele ser una manifestación relativamente común en otras variantes de epilepsia benigna de la infancia36,37, en estos casos es excepcional. No obstante, en esta serie se incluye a un varón que a los 6,9 años de edad consultó por haber presentado en vigilia dos episodios de pérdida de conciencia, de breve duración, distanciados en el tiempo por unos 3 meses, sin sintomatología motora y/o sensitiva y con paroxismos centrotemporales bilaterales. No se prescribieron fármacos anticonvulsivos y a lo largo de su control evolutivo de 5,8 años tan sólo volvió a presentar otro episodio de iguales características. También se incluyen dos varones que consultaron por presentar salvas de parpadeo pluricotidianas, sin alteración de la conciencia, y acompañados de forma esporádica por versión ocular y/o cefálica. En ambos casos se detectaron de manera reiterada y exclusiva paroxismos rolándicos interictales, con una respuesta terapéutica excelente y una adaptación escolar y psicosocial normales. En la mayoría de los pacientes las crisis tenían una duración breve; sin embargo, conviene resaltar que en algunos casos los episodios convulsivos son más prolongados, e incluso pueden manifestarse y/o iniciarse, tal como ocurrió en dos casos de esta serie, como un estado epiléptico10,20. La recuperación después de la somnolencia posictal fue completa y sin secuelas, salvo el caso de un paciente que presentó una hemiparesia posictal transitoria que a priori no debería excluir el diagnóstico de epilepsia rolándica20,38.
El diagnóstico de las epilepsias sigue siendo esencialmente clínico, pero en este caso el trazado electroencefalográfico es una prueba complementaria concluyente4,10,11,20–22. En estos pacientes el trazado interictal se caracteriza por una actividad basal bien estructurada con paroxismos caracterizados por una onda puntiaguda muy amplia seguida por una onda lenta de menor voltaje, que habitualmente agrupados o en racimo adquieren la forma de complejos punta-onda, localizados en la región centrotemporal. Son contralaterales a la manifestación clínica motora y, en proporción variable, se difunden a regiones adyacentes y/o al otro hemisferio cerebral de forma independiente o sincrónica. Esta actividad paroxística es muy persistente durante el registro en vigilia, y tiende a activarse durante el sueño, e incluso, tal como ocurrió en dos pacientes de esta serie, los paroxismos tan sólo aparecen durante el sueño. De tal modo que ante una anamnesis sugestiva de epilepsia rolándica y con trazados normales convendría repetir los estudios electroencefalográficos con privación de sueño4,20,23. No obstante, si bien la presencia de la actividad paroxística en la región rolándica es un elemento capital para el diagnóstico de esta entidad, su presencia no siempre es permanente ni se mantiene siempre en la misma localización; y como ocurría en esta serie, puede emigrar de un lado a otro, ser bilateral o acompañarse de focos en otras localizaciones. Además, convendría tener presente que estas manifestaciones electroencefalográficas también pueden aparecer ocasionalmente en niños que nunca presentarán crisis convulsivas, e incluso en pacientes con epilepsias sintomáticas4,20,23,39,40. Por tanto, para establecer el diagnóstico definitivo y clasificación sindrómica de estos pacientes habría que considerar conjuntamente el trazado electroencefalográfico y la secuencia semiología de las crisis y, desde luego, contar con un control evolutivo relativamente prolongado, tal como ocurría en esta serie, dado que las características diferenciales entre los distintos síndromes epilépticos pueden resultar difíciles especialmente al comienzo de la enfermedad. Fue precisamente el seguimiento continuado lo que permitió aseverar que en aquellos casos con semiología relativamente excepcional, como ocurría en dos pacientes con salvas de parpadeo y un tercero con síncopes ictales, se objetivaron de manera reiterada paroxismos centrotemporales bilaterales y/o con tendencia a generalizar, presentando una buena respuesta terapéutica en el caso de los primeros dos pacientes (13,2 y 7,0 años de evolución) y una remisión espontánea sin medicación en el caso del tercero (4,8 años de evolución).
La normalidad de los estudios de neuroimagen en la epilepsia rolándica ha sido considerada criterio diagnóstico4,11,23; sin embargo, y posiblemente en relación con la mayor capacidad resolutiva técnica actual, se han ido describiendo diferentes tipos de lesiones cerebrales subyacentes no evolutivas en estos pacientes que han sido consideradas hallazgos casuales, ya que generalmente no condicionan el curso y/o pronóstico de la enfermedad23,41, aunque según el caso podrían interferir en el desarrollo cognitivo e incluso tratarse de lesiones tumorales20,42,43. En esta serie, aunque los hallazgos neurorradiológicos (angioma venoso en lóbulo temporal derecho, y quiste subaracnoideo en fosa posterior) no condicionaron cambios evolutivos significativos, la experiencia acumulada hace considerar la conveniencia de realizar estudios de resonancia magnética cerebral en estos pacientes10.
Aunque está garantizada una excelente respuesta a los fármacos clásicos, siendo el valproato y la carbamacepina los de primera elección10,20,22,28, el ritmo nictameral, preferentemente nocturno, junto con la baja frecuencia y breve duración de las crisis, que en algunos casos harían discutible el diagnóstico sindrómico, y especialmente ante la posibilidad de un empeoramiento y/o generalización de las crisis, e incluso de evoluciones atípicas, inducidas por distintos fármacos anticonvulsivos44–47, hacen recomendable la abstención terapéutica. Su abordaje terapéutico debería ser similar al recomendado en las convulsiones febriles, es decir, administración domiciliaria de benzodiacepinas ante una crisis prolongada y, salvo múltiples recurrencias y/o ansiedad familiar, evitar el tratamiento médico continuado4. El pronóstico es muy favorable con una tendencia a la remisión espontánea en el curso de unos años, tal como se recoge en esta serie, y sin interferir en la integración escolar y/o social de los pacientes4,10,11,19,20. No obstante, algunos casos presentan evoluciones atípicas, tales como una actividad continua de punta-onda en sueño lento con signos de deterioro cognitivo y/o conductual o agnosia verbal auditiva adquirida con afasia o síndrome de Landau-Kleffner16,47,48 que condicionan el pronóstico, como sucedió en dos pacientes de esta serie. Diversos autores han cuestionado la benignidad de este síndrome epiléptico, ya que se ha detectado en pacientes con epilepsia rolándica una afectación cognitiva transitoria relacionada directamente con las descargas paroxísticas subclínicas que, al manifestarse en una época crítica del aprendizaje escolar, podría conllevar una evidente repercusión cognitiva12–16,49 que, posiblemente, sería susceptible de mejoría con fármacos antiepilépticos que redujeran la frecuencia de las descargas paroxísticas50. Para conocer la verdadera dimensión de esta eventualidad sería preceptivo incluir estudios neuropsicológicos seriados en los protocolos de valoración y seguimiento de los pacientes con epilepsia rolándica en orden a detectar o excluir dicha afectación cognitiva.
En suma, la epilepsia parcial benigna infantil con paroxismos centrotemporales o epilepsia rolándica constituye una patología específicamente pediátrica que afecta preferentemente a varones en edad escolar. Su secuencia semiológica es bastante característica y raramente acontece en otras epilepsias, y es imprescindible documentar paroxismos centrotemporales para confirmar el diagnóstico. Su pronóstico es excelente; sin embargo, dado que podrían cursar con una evolución atípica y/o una afectación cognitiva transitoria, sería conveniente mantener un riguroso control evolutivo de estos pacientes.
