




Caso clínico
Lactante de sexo masculino remitido a nuestro hospital desde otro centro a los 4 meses de edad por estatus epiléptico y pérdida de adquisiciones psicomotoras.
Entre sus antecedentes familiares destacaban: ausencia de consanguinidad; padre con paraparesia asimétrica atribuida a poliomielitis en su infancia; madre con migraña; hermana de la madre, acondroplasia; hermana de 8 años de edad sana. Fruto de una segunda gestación bien controlada de 37 semanas, con un peso al nacimiento de 2.800 g. El período neonatal cursó sin incidencias.
A los 2 meses y medio de vida, 5 días después de haber administrado la vacuna heptavalente, presentó un primer episodio crítico consistente en desconexión ambiental, versión ocular a la derecha, parpadeo izquierdo y movimientos de chupeteo. Ingresó en otro centro donde se refirió una exploración física normal. El electroencefalograma (EEG) mostró paroxismos de punta-onda en áreas temporales izquierdas y el estudio metabólico (estatus redox, aminoácidos plasmáticos) y la resonancia magnética (RM) cerebral resultaron normales. Siguió presentando múltiples crisis diarias de similares características pese a la administración de diferentes antiepilépticos (ácido valproico, oxcarbazepina, clonazepam, fenitoína y fenobarbital en diferentes combinaciones), y se evidenció la pérdida de las adquisiciones psicomotoras previamente conseguidas.
En el examen físico al ingreso en nuestro hospital destacan: palidez cutánea, hipopigmentación de mamilas y pelo escaso, áspero e hipopigmentado con cejas poco pobladas (fig. 1). No se observan rasgos dismórficos ni visceromegalias. La somatometría muestra un peso de 6 kg (P25), talla de 68 cm (P75) y perímetro craneal de 40 cm (2 DE). Se evidencia ausencia de contacto visual, disminución de movimientos espontáneos y marcada hipotonía axial con ausencia de sostén cefálico. El examen de fondo de ojo, pares craneales, fuerza y tono en extremidades es normal. Los reflejos osteotendinosos son vivos, sin clonías, con reflejo cutaneoplantar extensor. El resto de la exploración física es normal.
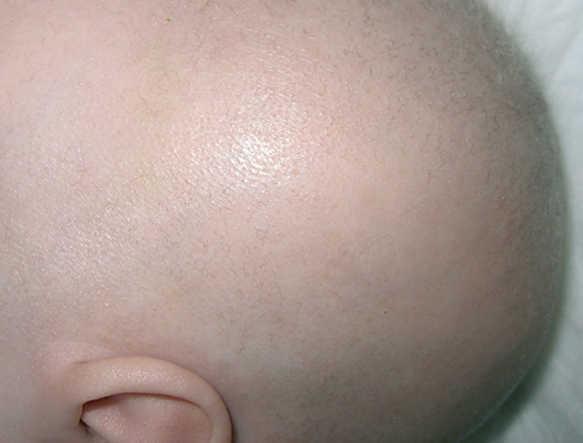
Figura 1. Aspecto macroscópico del pelo.
Se practican las siguientes exploraciones complementarias: hemograma, función renal y hepática, ionograma, albúmina y ácido úrico; estudio de líquido cefalorraquídeo (LCR) (leucocitos, glucosa, proteínas); estudio metabólico (estatus redox y aminoácidos en plasma y LCR, amonio y biotinidasa plasmática, y ácidos orgánicos en orina); electrorretinograma y potenciales evocados visuales y auditivos, todas ellas con resultados normales.
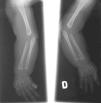
El EEG muestra una pobre estructuración del trazado de fondo, con múltiples descargas de punta-onda en ambas áreas temporales. En la serie esquelética destacan la reacción perióstica en huesos largos y una deformidad en copa de las metáfisis radiales (fig. 2). La densitometría ósea es normal. La RM y angiorresonancia cerebrales revelan un retraso de la mielinización, un infarto isquémico reciente en pálido derecho y unas arterias vertebrales elongadas y tortuosas (fig. 3).

Figura 2. Radiografía simple que muestra la reacción perióstica en huesos largos y una deformidad en copa de ambas metáfisis radiales.

Figura 3. Angiorresonancia cerebral que muestra el aspecto tortuoso de ambas arterias vertebrales.
Pregunta
1. ¿Cuál es su diagnóstico de presunción?
2. ¿Qué exámenes adicionales deben realizarse para confirmarlo?
Enfermedad de Menkes
La aparición de epilepsia refractaria y de deterioro neurológico en un lactante varón con hipopigmentación cutánea y las alteraciones del pelo descritas obligan a descartar la enfermedad de Menkes. Las anomalías de la angiorresonancia y de la serie esquelética son características de esta enfermedad. Además, el examen microscópico del pelo mostró la existencia de pili torti y se obtuvieron niveles extremadamente bajos de cupremia (15 μg/dl; valor normal: 70-140 μg/dl) y de ceruloplasmina sérica (7,23 mg/dl; valor normal: 19-45 mg/dl), datos que apoyan firmemente esta sospecha diagnóstica 1-3.
La enfermedad de Menkes es una enfermedad neurodegenerativa debida a mutaciones en el gen ATP7A, localizado en el cromosoma Xq12-q13 4-6, que codifica una ATPasa transportadora de cobre. Sigue un patrón de herencia recesivo ligado al cromosoma X que determina que los enfermos sean varones y que sus madres sean heterozigotas portadoras asintomáticas; excepcionalmente se describe la enfermedad también en el sexo femenino 7. Aproximadamente un tercio de los casos se debe a mutaciones de novo.
Las mutaciones en el gen ATP7A determinan una alteración del transporte del cobre a través de la membrana celular. La consecuente disminución de su biodisponibilidad impide su correcta utilización en la síntesis de ceruloplasmina, superóxido dismutasa y de muchas otras enzimas que contienen cobre, lo que da lugar a las numerosas manifestaciones clínicas asociadas a la enfermedad de Menkes. Las primeras manifestaciones suelen aparecer a los 2-3 meses de vida en forma de hipotermia, dificultad en la alimentación, pérdida de adquisiciones psicomotoras, convulsiones e hipotonía. El deterioro neurológico es habitualmente grave, con epilepsia intratable, y son características la elongación y tortuosidad de los vasos intracraneales y los hematomas subdurales. Las anomalías del pelo consisten, entre otras, en la existencia de pili torti, moniletrix y tricorrexis nudosa. La radiología simple muestra la presencia de numerosos huesos wormianos en calota, una osteoporosis generalizada y la reacción perióstica y ensanchamiento metafisario en huesos largos. Otras manifestaciones son hernias inguinales, diarrea, divertículos vesicales y la degeneración retiniana 2,3,8.
El diagnóstico es fácil en los pacientes con valores séricos bajos de cobre y ceruloplasmina y con manifestaciones clínicas y radiológicas típicas. Puede confirmarse por la demostración de la acumulación intracelular del cobre en el cultivo de fibroblastos y mediante el estudio molecular del gen ATP7A. Este último permite además la identificación de portadoras y el ulterior consejo genético 9.
La enfermedad de Menkes conduce a la muerte de la mayoría de los pacientes no tratados antes de los 3 años de edad. La administración parenteral de cobre es el único tratamiento que ha demostrado alguna eficacia y debe iniciarse tan pronto como se llegue al diagnóstico para evitar el deterioro neurológico progresivo. El tratamiento no corrige sin embargo las anomalías óseas o la hiperlaxitud del tejido conjuntivo, incluso en aquellos pacientes que evolucionan bien desde el punto de vista neurológico. De los diferentes preparados utilizados en la enfermedad de Menkes, el cobre-histidina es el que ha proporcionado mejores resultados. Se recomiendan dosis de entre 200 y 1.000 mg al día por vía subcutánea una vez al día o 2-3 veces por semana 8,10. Aunque permite corregir las concentraciones de cobre y ceruloplasmina séricas, la respuesta clínica es variable y depende de la precocidad del tratamiento y de la actividad residual de la ATPasa transportadora: se describen casos con buena evolución neurológica tras 17 años de seguimiento mientras que otros, con similares manifestaciones en el momento del diagnóstico, evolucionan de forma desfavorable y fallecen precozmente a pesar del inicio temprano del tratamiento 10.
