
La enteropatía pierde proteínas es la expresión clínica de varias enfermedades gastrointestinales y extradigestivas. Se caracteriza por una pérdida excesiva de proteínas por el intestino con una disminución de la albúmina sérica, que es consecuencia de la alteración del equilibrio entre la síntesis de proteínas en el hígado y la pérdida de las mismas por las heces1.
Inicialmente, la existencia de una hipoproteinemia sin proteinuria se atribuyó a una alteración de la síntesis hepática de proteínas. Más tarde, cuando se comprobó que la síntesis hepática no estaba alterada, se etiquetó de "hipoproteinemia idiopática".
Welch, en 1937, analizando las heces de pacientes con colitis ulcerosa, comprobó que excretaban más nitrógeno que las personas sanas y que no tenían esteatorrea. La hipoproteinemia de estos enfermos la atribuyó a una mayor pérdida de éstas por el colon. Diez años más tarde, Maimon (1947) comunica que los pacientes con enfermedad de Ménétrier pierden mucha proteína por los pliegues hipertrofiados del estómago y Kimble, Steinfeld y Citrin en 19572 hicieron las primeras descripciones del aumento de la secreción de albúmina en el estómago y de proteínas por el intestino en la colitis ulcerosa. Apartir de estas descripciones se han desarrollado numerosas técnicas para localizar el lugar de la lesión, valorar la cantidad de proteínas que se pierden en las diferentes enfermedades, entender mejor la patogénesis de esta pérdida proteica y comprobar la eficacia del tratamiento.
En la tabla 1se pueden ver las numerosas y variadas enfermedades que cursan con esta manifestación.

Métodos de estudio
Las proteínas séricas pueden identificarse en las secreciones gastrointestinales por métodos electroforéticos2, inmunológicos e inmunoelectroforéticos. Este tipo de estudios tiene el inconveniente de la dificultad de aspirar las secreciones gastrointestinales y que las proteínas pueden degradarse con la luz.
Las pruebas se dividen en dos grupos:
1.Determinación del aclaramiento fecal de alfa-1-antitripsina (a1-AT), una proteína que resiste el catabolismo de las proteasas en el intestino.
2.Cuantificación de la excreción fecal de una macromolécula radiactiva inyectada por vía intravenosa.
El aclaramiento fecal de a1-AT fue descrito en 1977 por Crossly y Elliot3, es la prueba más fidedigna para estudiar las pérdidas fecales de proteínas superando a la cuantificación aislada de proteínas. La a1-AT es una proteína con un peso molecular de 50.000, que resiste la proteólisis durante el tránsito intestinal y no se reabsorbe, cuando está presente en las heces indica que existe una pérdida de proteínas en algún segmento del tubo digestivo.
Floret et al4 demostraron que el estudio del aclaramiento de a1-AT era superior al simple análisis de la concentración fecal de proteínas y que tenía una buena correlación con el aclaramiento de albúmina en el intestino y la concentración en plasma de proteínas cuando éstas se marcaban con creatinina51; la sensibilidad, especificidad y el valor predictivo positivo era del 93,3, 90 y 97,7%, respectivamente, siendo el valor predictivo negativo del 75%.
El cálculo se realiza aplicando la siguiente fórmula:
Concentración fecal de a1-AT (mg/100g heces)*peso de heces
Concentración en suero de a1-AT (mg/dl)
Los resultados se expresan en miligramos por gramo de heces secas.
Existen variaciones individuales de hasta dos veces su concentración normal; no obstante, en los individuos que tienen una enteropatía pierde proteínas la eliminación puede ser hasta 10veces superior.
La prueba puede invalidarse en las siguientes circunstancias4:
Déficit grave de a1-AT.
Recién nacidos de una semana, por la elevada concentración de a1-AT en el meconio.
Contaminación de las heces por orina de pacientes con proteinuria.
En el síndrome de Zollinger-Ellison.
Cuando hay hemorragia intestinal.
La cuantificación de la excreción fecal de macromoléculas radiactivas inyectadas por vía intravenosa se utilizó para estudiar las pérdidas fecales de proteínas, pero tiene el inconveniente que se modifican en la luz y se reabsorben.
Se han empleado albúmina marcada con 131 I, albúmina y transferrina 51 Cr, albúmina con 99 mTc, ceruloplasmina 67 Cu, PVP 131I. Todos estos test han sido superados por el aclaramiento de a1-AT.
Fisiopatología
Para comprender la etiopatogenia de las alteraciones de los linfáticos es preciso hacer un breve recordatorio de su estructura y función.
El sistema linfático5 en su conjunto, está integrado por el tejido linfático y por los vasos linfáticos. El primero es un tejido disperso por el organismo y muy diferenciado desde el punto de vista funcional. Pertenecen al mismo los ganglios linfáticos, la pulpa blanca del bazo, las amígdalas y el tejido linfático de la mucosa del intestino y del aparato respiratorio, del timo, de la médula ósea y también los linfocitos contenidos en la sangre y en los tejidos. La otra parte del sistema linfático lo forman los vasos linfáticos que representan una vía accesoria por la que los líquidos de los espacios intersticiales pueden llegar a la sangre.
Los linfáticos pueden transportar proteínas e incluso partículas mayores fuera de los espacios tisulares. Estos no pueden pasar directamente por absorción hacia la sangre capilar y si no fuera por el drenaje linfático, las proteínas se acumularían en el espacio tisular y por su presión osmótica tenderían a retener cantidades crecientes de agua en los tejidos.
Los capilares linfáticos empiezan en fondos de saco ciegos y se unen para constituir vasos linfáticos de calibre mayor y de paredes gruesas. Todos acaban drenando en dos troncos linfáticos principales: el conducto torácico y la gran vena linfática. El conducto torácico se vacía en el sistema venoso en la unión de las venas subclavia y yugular interna derechas. En definitiva, toda la linfa recogida del cuerpo es devuelta al sistema circulatorio sanguíneo para mantener el contenido líquido de la sangre. La mayor parte de la linfa reunida por los linfáticos, antes de ser devuelta al sistema circulatorio sanguíneo, atraviesa una o varias estructuras pequeñas: los ganglios linfáticos.
El sistema linfático del aparato digestivo nace en los plexos mucosos, submucoso e intermuscular. Además de su función general, tiene la misión de transportar las grasas. Cada vellosidad del intestino delgado posee un diminuto conducto quilífero o linfático central que drena en los plexos.
Las proteínas no se pierden por la luz intestinal gracias a la integridad del epitelio que actúa como una barrera y por el adecuado drenaje de los vasos linfáticos. Cuando se altera alguno de estos mecanismos, las proteínas se pierden por el intestino y aparece el cuadro de enteropatía pierde proteínas. Esto puede ocurrir en tres situaciones: por obstrucción del drenaje linfático, por inflamación de la mucosa con ulceración de la misma o por inflamación de la mucosa sin ulceración.
La obstrucción primaria de los linfáticos (linfangiectasia intestinal6) es una afección congénita, caracterizada por una malformación linfática con dilatación de los quilíferos de las vellosidades y de los linfáticos subserosos. La obstrucción del drenaje linfático del intestino origina una rotura de los vasos quilíferos intestinales con salida de linfa a luz del intestino, hay una pérdida crónica de proteínas, de grasa, de linfocitos y de inmunoglobulinas en la luz intestinal y por las heces, produciendo hipoalbuminemia y esteatorrea.
En la etiopatogenia de la obstrucción de los linfáticos de origen cardíaco o vascular7, las proteínas se pierden por la existencia de un aumento de la resistencia al flujo de la linfa desde el conducto torácico a la circulación general por causa del incremento de la presión en el sistema venoso central, esto produce congestión de los sinusoides hepáticos y dilatación de los linfáticos intestinales similar a lo que acontece en la linfangiectasia intestinal8. Después de la intervención de Fontan9, la pérdida de proteínas o la alteración de la función hepática10 se produce por una alteración en el territorio venoso subdiafragmático11. Esta región está compuesta por dos tipos de circulaciones distintas dinámicamente: la esplácnica, que drena el tubo digestivo teniendo como resistencia añadida el hígado, y la sistémica, que drena las extremidades inferiores y los riñones. Las alteraciones del flujo esplácnico pueden explicar las complicaciones de origen gastrointestinal como la ascitis y pérdida de proteínas, que se producen por congestión en esta región12.
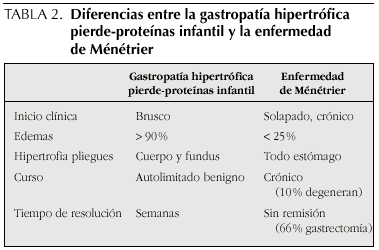
Si hay inflamación de la mucosa sin ulceración, la pérdida de proteínas se produce por aumento de la permeabilidad, por pérdida transcelular o por las uniones intercelulares13, como sucede en la enfermedad de Ménétrier. En la gastroenteritis eosinofílica la pérdida se produce por la inflamación de la mucosa y liberación de sustancias vasoactivas que aumentan la permeabilidad en los capilares y en el epitelio.
Cuando existe ulceración o erosiones de la mucosa e inflamación, la barrera epitelial desaparece y las macromoléculas del espacio intersticial difunden libremente hacia la luz del intestino. En esta situación coexisten cambios de la permeabilidad capilar y de la circulación linfática. Este es el mecanismo en la enfermedad inflamatoria intestinal.
Manifestaciones clínicas
Las enfermedades que asocian pérdida de proteínas por el intestino tienen unos síntomas y signos comunes: edemas, diarrea, vómitos, dolor abdominal, hipoproteinemia, hipoalbuminemia, hipogammaglobulinemia, linfopenia, con pérdida selectiva de linfocitos CD3 y CD4+, hipocalcemia y esteatorrea. En algunos casos es llamativo el retraso del desarrollo.
Tratamiento
El tratamiento es diferente según el mecanismo etiológico:
1.Dieta hipograsa con triglicéridos de cadena media, octreótido14 o anastomosis linfo-venosa en los casos de linfangiectasia intestinal primaria
2.Corticoides, inhibidores de la secreción ácida, anticuerpos monoclonales contra los receptores del factor de crecimiento epidermal15 en la enfermedad de Ménétrier
3.Diuréticos, digital e infusiones de albúmina como tratamiento sintomático, más corticoides16, heparina17, corrección quirúrgica o trasplante cardíaco en los casos secundarios cardiopatía con intervención tipo Fontan18.
