
La enfermedad de Addison o insuficiencia suprarrenal primaria es poco frecuente en la edad pediátrica y de diagnóstico a veces difícil por la inespecificidad y la presentación insidiosa de los síntomas, siendo la crisis suprarrenal una emergencia vital. Resulta necesario tener un alto índice de sospecha ante un cuadro clínico compatible para realizar un diagnóstico y tratamiento precoz.
Presentamos un estudio retrospectivo descriptivo de los datos epidemiológicos, clínicos y etiológicos de 5 pacientes diagnosticados de enfermedad de Addison en nuestro hospital. Los hallazgos clínicos más frecuentes al diagnóstico fueron la deshidratación, la hiponatremia y la hiperpigmentación cutánea. Los pacientes presentaron niveles deficientes de cortisol y anticuerpos antiadrenales positivos. El único paciente en el que los anticuerpos fueron negativos presentaba un síndrome poliglandular.
Addison's disease or primary adrenal insufficiency is a rare disease in children. The signs and symptoms at diagnosis are frequently non-specific and insidious. Since adrenal crisis represents an emergency, it is important to be aware and to have a high degree of suspicion of the disorder in order to achieve an early diagnosis and treatment.
We present a retrospective study describing the epidemiological, clinical and etiological data at diagnosis of five patients with Addison's disease followed up in our hospital. Dehydration, hyponatremia and skin hyperpigmentation were the most prevalent signs and symptoms at onset of the disease. The patients had low serum cortisol levels and positive adrenal antibodies. One patient with negative antibodies presented with a polyglandular syndrome.
La enfermedad de Addison (EA) consiste en una disfunción o hipofunción de la corteza suprarrenal, como consecuencia ocurre una producción insuficiente de glucocorticoides y mineralocorticoides, y una elevación secundaria de la hormona corticotropa (ACTH) y de la actividad de renina plasmática1. Su incidencia es de 0,8-1,4 casos por 100.000 habitantes/año2-4. Es poco frecuente en la edad pediátrica, pero potencialmente letal si no se diagnostica de forma precoz5,6.
La etiología de la insuficiencia suprarrenal primaria (ISP) puede ser adquirida o hereditaria. En adultos, más del 80% de los casos son causados por la destrucción autoinmunitaria de la glándula suprarrenal. La hiperplasia suprarrenal congénita (HSC) por déficit de la enzima 21-hidroxilasa es la causa más frecuente en niños1,2,4,7.
La EA de causa autoinmunitaria puede aparecer como una enfermedad aislada o formar parte de un síndrome poliglandular autoinmunitario (SPA)1,2,8-11. El SPA tipo 1 es poco frecuente, pero a veces se presenta en niños asociando ISP, hipoparatiroidismo y candidiasis mucocutánea crónica. Otras causas de ISP son las adquiridas (infección por tuberculosis, VIH, micosis, enfermedades malignas infiltrativas…) y las enfermedades hereditarias, como adrenoleucodistrofia ligada al cromosoma X, hipoplasia suprarrenal congénita (mutaciones en el gen DAX-1) o la deficiencia familiar de glucocorticoides4,8,12.
En esta revisión, se comunican 5 casos de EA al diagnóstico, con el objetivo de reforzar la importancia de la sospecha clínica del cuadro de insuficiencia suprarrenal ante la presencia de síntomas compatibles.
Pacientes y métodosSe presenta un estudio retrospectivo descriptivo, detallando los factores epidemiológicos, las formas de presentación clínica, la etiología y el tratamiento de 5 pacientes diagnosticados de EA en este centro durante los últimos 7 años. Los criterios utilizados para diagnosticar la EA fueron una historia clínica compatible y la confirmación de ISP mediante la demostración de niveles séricos bajos de cortisol y elevados de ACTH. En el estudio se excluyen los casos de HSC.
ResultadosTodos los pacientes son varones, con edades comprendidas entre los 3 y 11 años en el momento del diagnóstico (tabla 1).
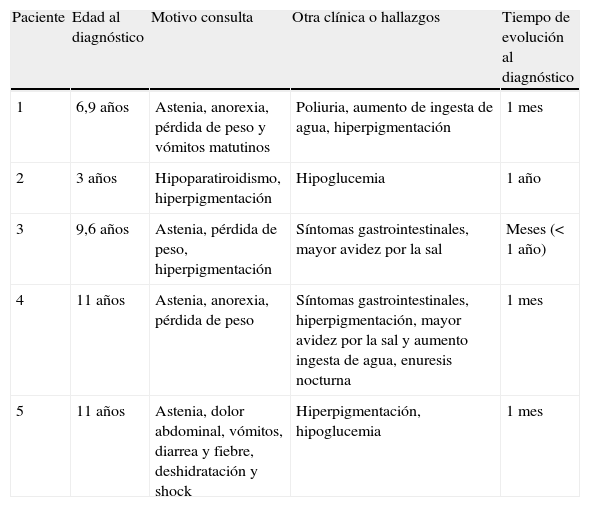
Formas de presentación clínica al diagnóstico de los pacientes
| Paciente | Edad al diagnóstico | Motivo consulta | Otra clínica o hallazgos | Tiempo de evolución al diagnóstico |
| 1 | 6,9 años | Astenia, anorexia, pérdida de peso y vómitos matutinos | Poliuria, aumento de ingesta de agua, hiperpigmentación | 1 mes |
| 2 | 3 años | Hipoparatiroidismo, hiperpigmentación | Hipoglucemia | 1 año |
| 3 | 9,6 años | Astenia, pérdida de peso, hiperpigmentación | Síntomas gastrointestinales, mayor avidez por la sal | Meses (< 1 año) |
| 4 | 11 años | Astenia, anorexia, pérdida de peso | Síntomas gastrointestinales, hiperpigmentación, mayor avidez por la sal y aumento ingesta de agua, enuresis nocturna | 1 mes |
| 5 | 11 años | Astenia, dolor abdominal, vómitos, diarrea y fiebre, deshidratación y shock | Hiperpigmentación, hipoglucemia | 1 mes |
SPA: síndrome poliglandular autoinmunitario.
Como antecedentes personales, un paciente presentó un síndrome de hiperactividad y déficit de atención (paciente 1) y otro paciente trombopenia, ptosis palpebral derecha e ingresos por acidosis metabólica, hipoglucemia e hiponatremia en el contexto de vómitos (paciente 2). Entre los antecedentes familiares, cabe destacar que la madre del paciente 3 presentaba una EA de origen autoinmunitario.
Los signos y los síntomas de presentación que se encontraron entre nuestros pacientes se resumen en la tabla 1. Los más frecuentes fueron la hiperpigmentación cutánea (100% de los casos) y la hiponatremia (en 4 de los 5 pacientes). El paciente 5 precisó ingreso en la Unidad de Cuidados Intensivos por presentar un cuadro de deshidratación y acidosis metabólica grave, que evolucionó a shock hipovolémico refractario e isquemia mesentérica aguda.
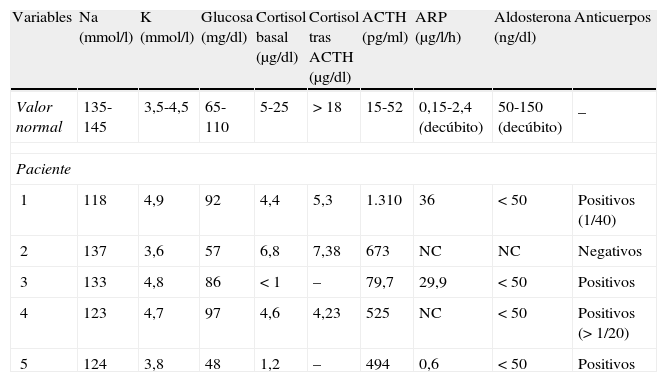
La sospecha diagnóstica de insuficiencia suprarrenal se confirmó en todos los casos mediante la demostración de niveles disminuidos de cortisol plasmático: 2 pacientes tuvieron niveles basales de cortisol claramente deficientes y en los otros 3 la prueba de estímulo con ACTH a dosis de 250 μg i.v. mostró una mínima e inadecuada elevación de los niveles de cortisol tras el estímulo. Todos los pacientes presentaron niveles elevados de ACTH, lo que confirmó el origen primario de la insuficiencia suprarrenal. Los hallazgos analíticos en el comienzo de la enfermedad se recogen en la tabla 2.
Hallazgos analíticos al diagnóstico de la enfermedad
| Variables | Na (mmol/l) | K (mmol/l) | Glucosa (mg/dl) | Cortisol basal (μg/dl) | Cortisol tras ACTH (μg/dl) | ACTH (pg/ml) | ARP (μg/l/h) | Aldosterona (ng/dl) | Anticuerpos |
| Valor normal | 135-145 | 3,5-4,5 | 65-110 | 5-25 | > 18 | 15-52 | 0,15-2,4 (decúbito) | 50-150 (decúbito) | _ |
| Paciente | |||||||||
| 1 | 118 | 4,9 | 92 | 4,4 | 5,3 | 1.310 | 36 | < 50 | Positivos (1/40) |
| 2 | 137 | 3,6 | 57 | 6,8 | 7,38 | 673 | NC | NC | Negativos |
| 3 | 133 | 4,8 | 86 | < 1 | – | 79,7 | 29,9 | < 50 | Positivos |
| 4 | 123 | 4,7 | 97 | 4,6 | 4,23 | 525 | NC | < 50 | Positivos (> 1/20) |
| 5 | 124 | 3,8 | 48 | 1,2 | – | 494 | 0,6 | < 50 | Positivos |
ARP: actividad de renina plasmática; NC: no consta realizado al diagnóstico.
En cuanto al diagnóstico etiológico, 4 de los 5 pacientes presentaron anticuerpos antiadrenales positivos. El único con anticuerpos negativos (paciente 2) asociaba trombopenia autoinmunitaria y el estudio de EA se realizó tras detectar un hipoparatiroidismo y candidiasis oral. El estudio genético-molecular del gen AIRE (autoinmune regulator gene) fue negativo y recientemente ha presentado temblor distal y la resonancia magnética craneal realizada muestra lesiones en la sustancia blanca que sugieren el diagnóstico de síndrome de Kearns-Sayre frente al de SPA13.
El estudio de ácidos grasos de cadena larga, los resultados para la prueba de la tuberculina y la serología para VIH fueron negativos en los casos estudiados.
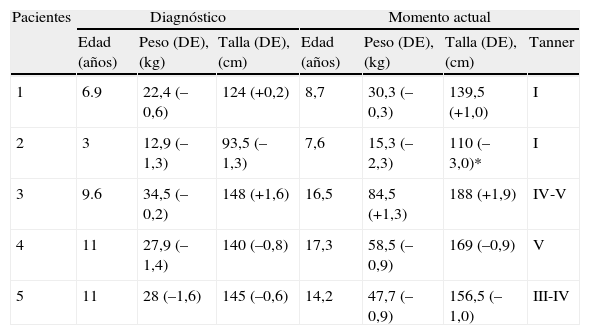
Los pacientes recibieron tratamiento durante la crisis suprarrenal con hidrocortisona i.v, siendo transferidos a tratamiento oral con hidrocortisona a dosis sustitutiva y fludrocortisona cuando la situación clínica lo permitió. La evolución ha sido favorable y presentan un crecimiento y desarrollo normal (tabla 3).
Medidas antropométricas y evolución
| Pacientes | Diagnóstico | Momento actual | |||||
| Edad (años) | Peso (DE), (kg) | Talla (DE), (cm) | Edad (años) | Peso (DE), (kg) | Talla (DE), (cm) | Tanner | |
| 1 | 6.9 | 22,4 (–0,6) | 124 (+0,2) | 8,7 | 30,3 (–0,3) | 139,5 (+1,0) | I |
| 2 | 3 | 12,9 (–1,3) | 93,5 (–1,3) | 7,6 | 15,3 (–2,3) | 110 (–3,0)* | I |
| 3 | 9.6 | 34,5 (–0,2) | 148 (+1,6) | 16,5 | 84,5 (+1,3) | 188 (+1,9) | IV-V |
| 4 | 11 | 27,9 (–1,4) | 140 (–0,8) | 17,3 | 58,5 (–0,9) | 169 (–0,9) | V |
| 5 | 11 | 28 (–1,6) | 145 (–0,6) | 14,2 | 47,7 (–0,9) | 156,5 (–1,0) | III-IV |
*Paciente 2: probable diagnóstico de síndrome Kearns-Sayre que asocia, entre otros, talla baja.
Los pacientes 1, 2 y 4 pertenecen a familias con talla media; el paciente 3 pertenece a familia con talla alta; el paciente 5 pertenece a familia con talla media-baja.
Todos los pacientes tienen en la actualidad edad ósea acorde a la edad cronológica, excepto los pacientes 2 y 5 que presentan un retraso de edad ósea de 2,5 años y 1 año respectivamente.
La EA es una entidad clínica potencialmente letal cuando su diagnóstico no se sospecha o se demora5,6. Los síntomas y signos se presentan generalmente de forma gradual pero a veces se manifiestan de forma brusca como insuficiencia suprarrenal aguda tras una situación de estrés (infección, trauma o intervención quirúrgica).
Los síntomas iniciales pueden ser astenia, pérdida de peso, vómitos o dolor abdominal, siendo estos comunes a muchas enfermedades. La presencia de avidez por la sal, deshidratación con hiponatremia e hiperpotasemia y, sobre todo, la hiperpigmentación cutánea nos debe hacer sospechar la enfermedad2,12,14.
Las formas de presentación clínica de nuestros pacientes fueron diversas, con un abanico que abarca desde un paciente que se diagnosticó como consecuencia de un estudio por hipoparatiroidismo y antecedentes de ingresos por deshidratación hasta un niño que presentó shock hipovolémico refractario a tratamiento. La hiperpigmentación cutánea estuvo presente en los 5 pacientes en mayor o menor grado y fue crucial para establecer el diagnóstico de sospecha.
En nuestra serie, todos los pacientes presentaron niveles séricos de cortisol y aldosterona disminuidos y de ACTH elevados. La presencia de clínica compatible junto con la determinación de electrolitos en sangre y el hallazgo de niveles disminuidos de cortisol sérico a primera hora de la mañana (< 3μg/dl o<5μg/dl durante la crisis suprarrenal) hacen el diagnóstico. Cuando el diagnóstico es dudoso es necesario realizar un test de estímulo con ACTH sintética (250μg i.v), considerándose la respuesta patológica cuando tras 60 min de su administración el pico de cortisol sérico es<18μg/dl8,10. El hallazgo de valores elevados de ACTH plasmática y de la actividad de renina plasmática con niveles relativamente bajos de aldosterona contribuyen a confirmar el diagnóstico de ISP.
Realizado el diagnóstico funcional de la ISP, es obligatorio investigar la etiología del proceso sin retrasar el inicio del tratamiento. Los anticuerpos antiadrenales son marcadores de adrenalitis autoinmunitaria, siendo muy sensibles y específicos para el diagnóstico de EA15. Detectamos anticuerpos antiadrenales positivos en 4 de los 5 pacientes comunicados, siendo negativos en un paciente con afección poliglandular y neuromuscular asociada. El hallazgo de anticuerpos antiadrenales positivos estableció la etiología autoinmunitaria en esos 4 pacientes.
Dada la alta frecuencia de asociación de ISP a otras endocrinopatías, es necesario completar el cribado de otros trastornos autoinmunitarios en todos los pacientes con EA. En los casos expuestos no observamos positividad de otros anticuerpos.
Otra prueba que se debe realizar en los pacientes con ISP es la determinación de ácidos grasos de cadena muy larga en sangre y orina para descartar adrenoleucodistrofia, especialmente en niños varones, si existen alteraciones neurológicas o del comportamiento o antecedentes familiares16,17. Los resultados de esta prueba fueron negativos en nuestros pacientes.
El inicio precoz del tratamiento sustitutivo con corticoides es fundamental para evitar la evolución tórpida y un desenlace fatal de la enfermedad y se debe acompañar de la administración de líquidos y electrólitos i.v. para corregir el desequilibrio ácido-base y normalizar los iones. Las dosis recomendadas de hidrocortisona son de 50 a 75mg/m2 de superficie corporal i.v. como bolo inicial, seguido de 75 a 100mg/m2 al día, repartido en 3 o 4 dosis. El tratamiento a largo plazo requiere la sustitución de glucocorticoides y mineralocorticoides. La hidrocortisona es el corticoide de elección por tener vida media corta e influir mínimamente en el crecimiento. La dosis que se utiliza es de 8-20mg/m2/día según edades y se administra distribuida en 3 dosis diarias, por vía oral. La deficiencia de mineralocorticoides se trata con fludrocortisona, a dosis de 0,05 a 0,2mg/día4,13. En situaciones de enfermedad o estrés, es necesario duplicar o triplicar la dosis por vía oral de hidrocortisona y si existe mayor gravedad, tratar con hidrocortisona i.m. o i.v. en un centro hospitalario18.
En conclusión, la insuficiencia suprarrenal es un trastorno poco frecuente en niños, que se presenta con síntomas inespecíficos. Se debe considerar su diagnóstico ante un cuadro clínico compatible, ya que la crisis suprarrenal representa una emergencia vital. Por lo tanto, el reconocimiento precoz y el tratamiento inmediato de la insuficiencia suprarrenal son críticos para la supervivencia de estos pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
