


La encefalopatía aguda necrosante (EAN) es una encefalopatía rápidamente progresiva que se presenta en niños por lo demás normales asociada a infecciones víricas comunes, como influenza y parainfluenza. La mayoría de los casos son esporádicos y no recurrentes, y se han descrito en pacientes asiáticos, aunque también se han comunicado casos aislados en países occidentales.
Recientemente se han encontrado mutaciones en heterocigosis de un gen que codifica una proteína componente del poro nuclear, denominada Ran Binding Protein 2 (RANBP2), en un número significativo de pacientes con esta forma familiar o recurrente de EAN.
A continuación se describe una familia española con la forma familiar y recurrente de EAN, en la que el estudio de secuenciación del gen RANBP2 fue negativo. Las mutaciones del gen RANBP2 no son el único alelo que determina susceptibilidad para el desarrollo de la forma familiar o recurrente de EAN.
Acute necrotizing encephalopathy (ANE) presents in children after common viral infections. Most cases of ANE are non-familial and non-recurrent and have been mainly reported in Asian patients, although ANE affects children worldwide. Recently, missense mutations in the gene encoding the nuclear pore protein Ran Binding Protein 2 (RANBP2) have been found in several families with familial or recurrent cases of ANE. We describe a Spanish family with familial and recurrent ANE without mutations in RANBP2. Mutations in RANBP2 are not the sole susceptibility alleles for familial or recurrent ANE.
La encefalopatía aguda necrosante (EAN) es una encefalopatía rápidamente progresiva que se presenta en niños por lo demás normales asociada a infecciones víricas comunes, como influenza y parainfluenza1. La mayoría de los casos son esporádicos y no recurrentes, y se han descrito en pacientes asiáticos, aunque también se han comunicado algunos casos aislados en países occidentales2,3. Se describió la forma familiar o recurrente de EAN con un patrón de herencia autosómico dominante de penetrancia incompleta en el año 2003 en una familia americana de origen europeo4. Posteriormente se identificó, a partir de estudios genéticos realizados en esa familia, su locus en 2q12.1-2q135. En 2009 se ha comunicado el hallazgo de mutaciones en heterocigosis de un gen que codifica una proteína componente del poro nuclear denominada Ran Binding Protein 2 (RANBP2) en todos los miembros afectados y portadores obligados de la familia original, así como en otras 11 familias adicionales con EAN familiar o recurrente6. Sin embargo, no se encontraron mutaciones del gen RANBP2 en otras 4 familias con EAN familiar o recurrente estudiadas; una de ellas corresponde a la familia que se presenta en este estudio.
Pacientes y métodosEl diagnóstico de EAN se basó en los 3 criterios siguientes6: a) el desarrollo de una encefalopatía rápidamente progresiva de inicio de uno a 3 días tras el comienzo de una enfermedad febril; b) lesiones simétricas multifocales en el estudio de resonancia magnética (RM) craneal con afectación de tálamos; se consideraron las lesiones de tronco del encéfalo como apoyo al diagnóstico, y c) exclusión, tanto a través de la clínica como de exámenes de laboratorio, de meningitis, encefalitis, errores congénitos del metabolismo y exposición a toxinas. La forma familiar o recurrente de EAN requirió de al menos uno de los siguientes criterios adicionales: a) un familiar con EAN confirmada; b) un familiar con historia de disfunción del sistema nervioso central inexplicada, de aparición en el contexto de enfermedad febril concurrente o infección reciente, o c) recurrencia de EAN en el caso índice.
Se obtuvieron muestras de 6 miembros de la familia estudiada que cumplían criterios de EAN familiar o de posible portador asintomático del trastorno. Previamente, se obtuvo consentimiento informado de los pacientes adultos y de los padres de los niños estudiados. Se secuenció el gen RANBP2 en búsqueda de mutaciones, tal como se ha descrito previamente6.
ResultadosEsta familia está constituida por un caso índice en el que se desarrollaron 2 episodios de encefalopatía desencadenados por infecciones (falleció en el segundo de los episodios), y por 5 familiares de segundo o tercer grado en los que se desarrolló encefalopatía concurrente con infecciones (de éstos fallecieron 3) (fig. 1, tabla 1).
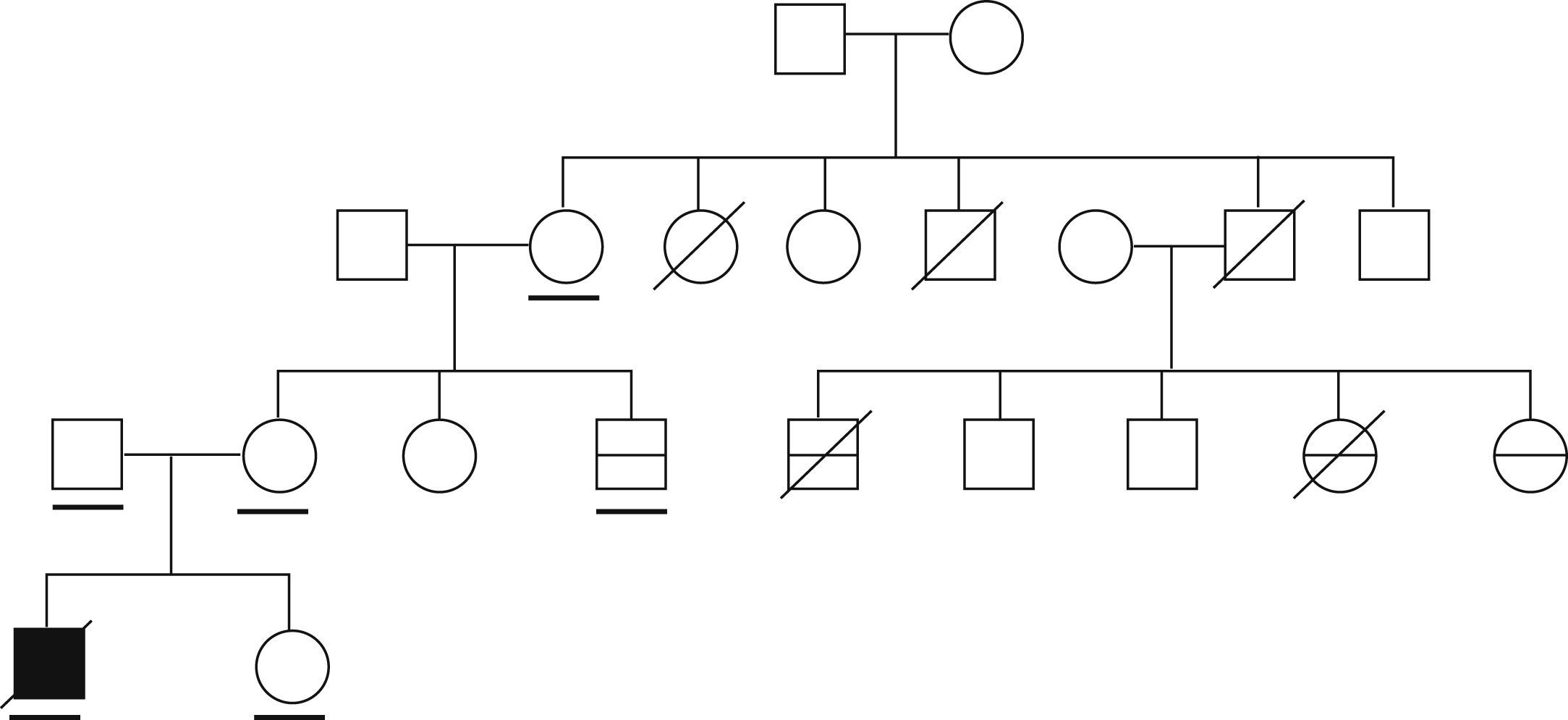
Características clínicas de los pacientes con encefalopatía aguda necrosante familiar y posibles portadores del trastorno
| Sujeto | Sexo/año de nacimiento | Características clínicas |
| II-1 | M/1940 | Portadora obligada |
| II-2 | M/ND | Caso posible. Fallecimiento atribuido a infección por influenza a la edad de 24 años (agudo, en horas) |
| II-4 | V/ND | Fallecimiento a la edad de 8 meses, causa desconocida |
| II-5 | V/ND | Portador obligado. Fallecimiento a los 60 años por hepatopatía |
| III-1 | M/1968 | Portadora obligada. Madre del propositus |
| III-3 | V/1977 | Caso probable. Encefalopatía a la edad de 3 años. Recuperación sin secuelas |
| III-4 | V/1971 | Caso probable. Encefalopatía a la edad de 15 meses con desenlace fatal |
| III-7 | M/1978 | Caso probable. Encefalopatía a la edad de 10 meses con desenlace fatal |
| III-8 | M/1980 | Caso posible. A la edad de 10 meses presentó convulsión afebril generalizada. Encefalitis atribuida a infección por varicela a la edad de 2 años (LCR: 20 células, proteínas elevadas: 1,25gr/dl). Posteriormente, epilepsia focal (temporal) sintomática remota |
| IV-1 | V/2005 | Caso índice |
LCR: líquido cefalorraquídeo; M: mujer; ND: no disponible; V: varón.
Primer episodio de encefalopatía a la edad de 3 meses:
Lactante previamente sano. Fiebre durante 24h, tos y rinorrea. Tras un día afebril presentó vómitos, irritabilidad, somnolencia y rigidez en extensión periódica. En la exploración física destacaron temblor, hipertonía de miembros e hiperreflexia. Se observaron deterioro progresivo del nivel de consciencia hasta el coma, una convulsión clónica generalizada y mioclonías generalizadas frecuentes.
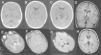
El análisis del líquido cefalorraquídeo (LCR) mostró proteínas de 1.192mg/dl, glucosa normal y 5 células (al día siguiente, las proteínas eran de 1.400mg/dl). La tomografía computarizada (TC) craneal mostró hipoatenuación de ambos tálamos y mesencéfalo (fig. 2). El electroencefalograma (EEG) durante las primeras 24h de la encefalopatía mostró discreto enlentecimiento de la actividad de fondo, sin actividad paroxística. Las proteínas totales en suero eran de 5,9g/dl el día del ingreso (6,6 a 8,7); de 5,4g/dl al día siguiente y, tras una semana, de 5,1g/dl. La alanina-aminotransferasa (ALT) era de 53 (5 a 45)U/l. Se inició tratamiento con vitaminas B1, B2, B6, biotina, B12 y carnitina.
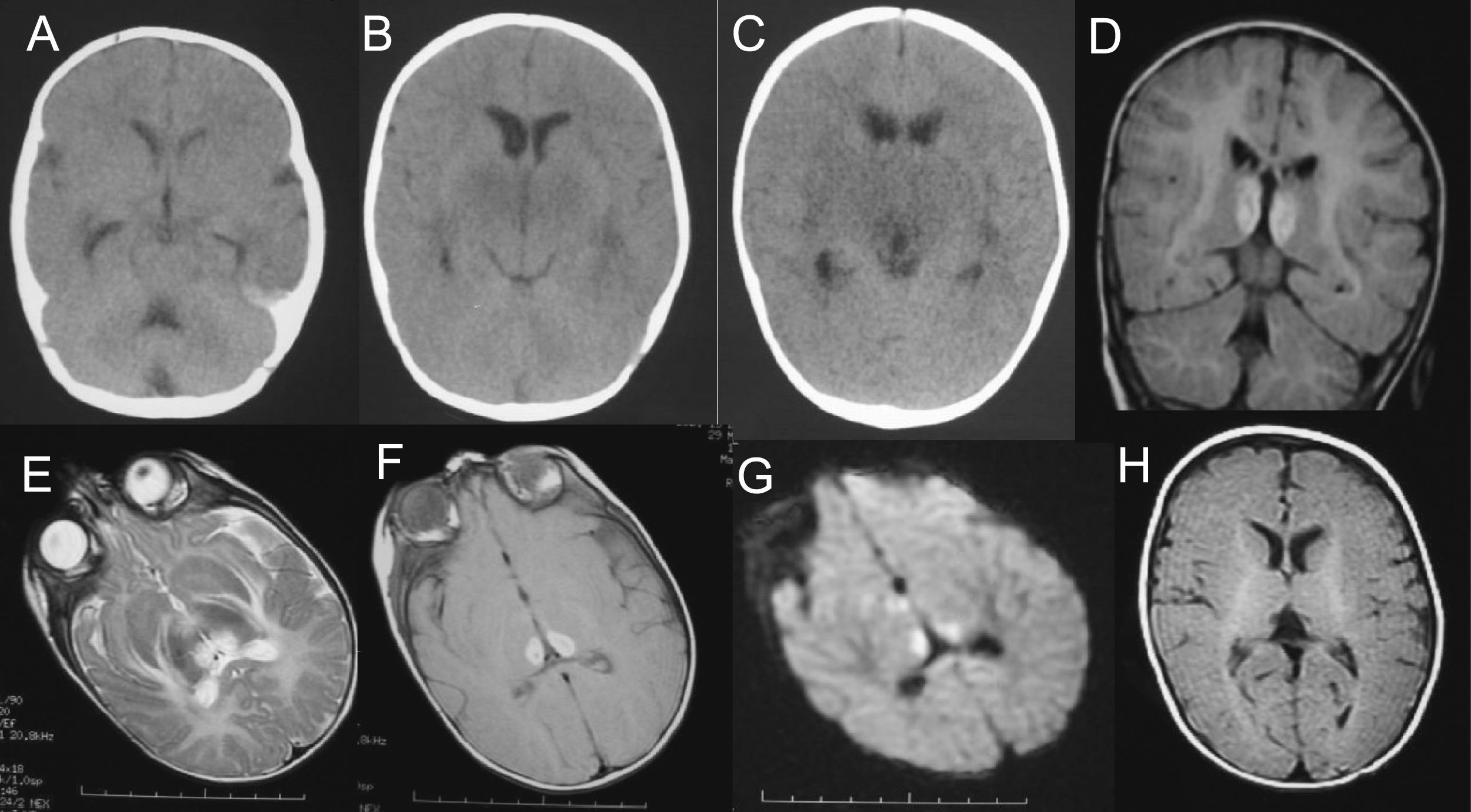
A-C) Tomografía computarizada craneal (durante las primeras 24h): hipoatenuación de los ganglios basales, tálamos y mesencéfalo, más marcada en sustancia gris periacueductal, junto con dilatación de astas temporales de ventrículos laterales y acueducto de Silvio. D-G) Resonancia magnética (RM) craneal a los 3 días de ingreso: se aprecia hiperintensidad de la señal en ambos tálamos posteromediales en secuencia ponderada en T1, T2, DP y en secuencia de difusión. No hay dilatación del sistema ventricular ni alteración de la señal en ganglios basales ni mesencéfalo. H) RM craneal realizada 6 semanas después del episodio de encefalopatía, imagen axial en secuencia ponderada en T1 a la altura de los ganglios de la base y los tálamos: resolución de las alteraciones previamente descritas. Estudio sin hallazgos de significado patológico.
Después de 3 días, el nivel de consciencia era normal y no presentaba focalidad neurológica. Una RM craneal en ese momento mostró hiperintensidad de la señal en ambos tálamos posteromediales en las secuencias T1, T2, DP, FLAIR y difusión (fig. 2). Se excluyó trombosis de venas cerebrales profundas y de senos durales mediante angiografía por RM.
Una semana después del ingreso, la situación clínica era de normalidad neurológica. El análisis citoquímico del LCR mostró 2 células, proteínas de 59mg/dl (0 a 40) y glucosa normal.
Tras este episodio de encefalopatía, el lactante se recuperó aparentemente sin secuelas. Una segunda RM craneal realizada 6 semanas después del episodio de encefalopatía fue normal.
Los siguientes estudios con muestras obtenidas durante el primer episodio de encefalopatía fueron normales: hemograma, bioquímica de orina y sedimento, electrolitos séricos, función hepática y renal, coagulación, amonio y lactato en varias determinaciones, piruvato, equilibrio acidobásico, aminoácidos en plasma y LCR, ácidos orgánicos en orina y sulfitest; en el LCR se estudiaron bandas oligoclonales, índice de inmunoglobulina G y reacción en cadena de la polimerasa (PCR) para virus del herpes; se realizaron diversas serologías en suero y LCR, valoración cardiológica y oftalmológica, ecografía abdominal, electromiograma, electroneurograma, potenciales evocados auditivos, isoformas de sialotransferrinas, perfil de acilcarnitinas y estudios del metabolismo energético en fibroblastos mediante técnicas de oxidación de sustratos energéticos con isótopos reactivos.
Segundo episodio de encefalopatía a la edad de 7 meses:
Comenzó con fiebre de 38 a 38,5°C junto a pérdida de apetito durante 2 días. Al final del segundo día presentó crisis focales de los miembros izquierdos, vómitos y obnubilación progresiva hasta el coma. Junto con esto, presentó mioclonías con estímulos acústicos, hiperreflexia y fontanela a tensión. Tras unas horas, tenía signos de herniación cerebral: postura en descerebración, midriasis arreactiva, hipertensión arterial, flacidez generalizada y finalmente falleció. El hemograma, la bioquímica de orina, los electrolitos, el equilibrio acidobásico, la función hepática y renal eran normales. Al ingreso, las proteínas totales en suero eran de 6,4g/dl (6,6 a 8,7), al segundo día eran de 5,6g/dl. El análisis del LCR mostró 20 linfocitos, 50 hematíes, proteínas de 317mg/dl, glucosa normal y PCR de virus del herpes negativa. El EEG mostró enlentecimiento de la actividad de fondo. El segundo día, horas antes del fallecimiento, la TC craneal mostró pérdida de diferenciación entre sustancia gris y sustancia blanca del diencéfalo y de ambos hemisferios cerebrales, junto con hipoatenuación de ambas regiones subinsulares.
Paciente III-3Tres años de edad. Tras 2 días de fiebre, tos y rinorrea presentó cefalea y vómitos durante 2 días adicionales. Al ingreso, el cuarto día, se observaron ataxia, nistagmo rotatorio vertical y horizontal, crisis mioclónicas, obnubilación e hiperreflexia. El análisis del LCR mostró 6 células y proteínas de 230mg/dl. El hemograma, el equilibrio acidobásico, la bioquímica de orina, los electrolitos y la función renal eran normales. El EEG mostró enlentecimiento de la actividad de fondo. Tras 2 a 3 días, la evolución fue muy satisfactoria. Diescisiete días después, la TC craneal era normal. El paciente tuvo recuperación sin secuelas. A la edad de 29 años, la RM craneal fue normal (realizada tras el diagnóstico del caso índice).
Paciente III-7A la edad de 10 meses presentó fiebre baja, pérdida de apetito y vómitos. Recibió ácido acetilsalicílico. Unas horas más tarde, el paciente presentó irritabilidad y se desarrolló en él un estado epiléptico focal con fiebre. Se observó deterioro de la consciencia hasta el coma y parada cardiorrespiratoria; finalmente falleció. El hemograma, la bioquímica de orina, los electrolitos y la función renal eran normales. El análisis del LCR mostró 8 células, glucosa de 82mg/dl y proteínas de Pandy 1+. La autopsia reveló edema cerebral y pulmonar.
DiscusiónLa forma familiar o recurrente de EAN cursa con episodios de encefalopatía aguda desencadenados por fiebre, habitualmente entre los 8 meses y los 6 años de vida, aunque se ha descrito el desarrollo de episodios de encefalopatía también en adultos de forma excepcional. Durante los episodios agudos, la RM detecta hiperintensidades en tálamos y troncoencéfalo en secuencias T2 y FLAIR. Junto con esto pueden encontrarse hiperproteinorraquia y elevación de las aminotransferasas como únicos marcadores bioquímicos de la encefalopatía4. El pronóstico de los episodios comprende desde la recuperación completa, hasta secuelas neurológicas permanentes (espasticidad, retraso mental, epilepsia) y fallecimiento. El 50% de los pacientes que sobreviven presenta una recurrencia que suele ser más grave que el episodio inicial. Los portadores obligados no siempre muestran las características clínicas. Esta entidad muestra semejanzas con el síndrome de Leigh y especialmente con la EAN esporádica, así como con otras encefalopatías de la infancia asociadas a infecciones víricas. Estudios del metabolismo energético realizados en miembros de la familia originalmente descrita mostraron un acoplamiento laxo de la fosforilación oxidativa en uno de los pacientes, por lo que se pensó inicialmente en una disfunción mitocondrial como posible origen de la enfermedad4. Sin embargo, el hallazgo de mutaciones missense del gen RANBP2, que codifica una proteína constituyente del poro nuclear en todos los afectados y portadores obligados de la familia original, así como en 11 de otras 15 familias adicionales con EAN familiar o recurrente estudiadas, abre nuevas vías a la investigación de la etiopatogenia de esta entidad6. El gen RANBP2 no es el único gen determinante de susceptibilidad para el desarrollo de la forma familiar o recurrente de EAN, lo que se deduce por el hecho de que no se encontraron mutaciones en 4 de las 16 familias estudiadas con este trastorno6. Por otro lado, es difícil especular acerca de los posibles mecanismos patogénicos del efecto de las mutaciones de este gen que codifica una proteína de 385kD localizada en la superficie citoplásmica del poro nuclear. Esta proteína se ha detectado asociada a microtúbulos7 y a las mitocondrias8,9 en las neuronas, lo que indica que desempeña un papel en el tráfico intracelular y en el mantenimiento de la energía. También parece desarrollar importantes funciones en la rotura de la membrana nuclear durante la mitosis10.
Se describió la primera familia española afectada por la forma familiar y recurrente de EAN, y se secuenció en varios miembros el gen RANBP2. Es recomendable que se considere este diagnóstico en pacientes con EAN recurrente o con EAN e historia familiar de síntomas neurológicos de aparición en el contexto de una infección, así como que se continúe investigando en la etiología genética de la forma familiar o recurrente de EAN a través del análisis de familias adicionales con este trastorno recientemente descrito.
FinanciaciónEste trabajo se ha financiado parcialmente mediante una beca del US National Institutes of Health NINDS K08NS050331.
