



El diagnóstico de la hipoplasia de cartílago-pelo o condrodisplasia metafisaria tipo McKusick (MIM 250250), se establece por talla baja disarmónica, fémur y tibia incurvados, falanges cortas y engrosadas, laxitud de ligamentos con característica limitación a la extensión de los codos y pelo ralo, inicialmente descrito en población amish estadounidense1. Es debido a mutaciones en el gen RMRP (MIM 157660; 9p13)2: gen nuclear no codificante que transcribe para una cadena de ARN que forma parte del complejo mitocondrial-RNasa-RMP participando en el ensamblaje de los ribosomas y en la progresión del ciclo celular. Con patrón de herencia recesiva, la severidad de las mutaciones da lugar a una clínica muy variada (de más leve a más grave): displasia metafisaria sin hipotricosis, hipoplasia de cartílago-pelo y displasia anauxética3.
Caso 1Varón de 7 meses, de padres sanos no consanguíneos españoles. Nació a las 40 semanas con 3.200g, 45cm y PC de 36cm. Ingresó a los 4 meses por gastroenteritis aguda. A la exploración presenta pelo ralo, escaso y frágil, acortamiento de extremidades y fémures algo incurvados en varo, manos con dedos cortos. Talla 65,5cm (−1,31DE) y peso 7.850g (−0,72DE). Estudio óseo: acortamiento de los huesos largos y ensanchamiento metafisario. Estudio inmunitario: leucopenia con neutropenia moderada y linfopenia (linfocitos T CD4 y B+ disminuidos); niveles disminuidos de IgA siendo normales los de IgM e IgG. Con estos datos se le diagnosticó clínicamente de hipoplasia de pelo-cartílago. A la edad de 3 años se solicita el estudio del gen RMRP revelando la presencia de 2 mutaciones en heterocigosis [g.236A>G, paterno; g.260G>A, materno]. Este diagnóstico permitió el desarrollo de una sonda para detectar la mutación paterna en el ADN fetal en sangre materna (técnica prenatal no invasiva) a las semanas de gestación 8 y 10 en 2 gestaciones posteriores de esta pareja. En la primera gestación se detectó la mutación paterna en el ADN fetal en sangre materna confirmado por amniocentesis (técnica invasiva) resultando el feto afecto. Se decidió la interrupción del embarazo. En la segunda gestación no se detectó la mutación paterna y el estudio prenatal invasivo lo confirmó (feto sano no portador; publicación en proceso). A los 9 años continúa con talla baja (108cm, [−4,06DE], peso 20kg [−2,25DE]), incurvamiento en varo de las extremidades moderada con seguimiento en rehabilitación. Pelo ralo. Ha presentado 2 neumonías sin identificación de gérmenes. Está correctamente vacunado según el calendario de su comunidad, presentando reacciones febriles leves con las vacunas de virus vivos. Toma trimetroprim sulfametoxazol de manera profiláctica 3 días en semana (fig. 1).
Caso 2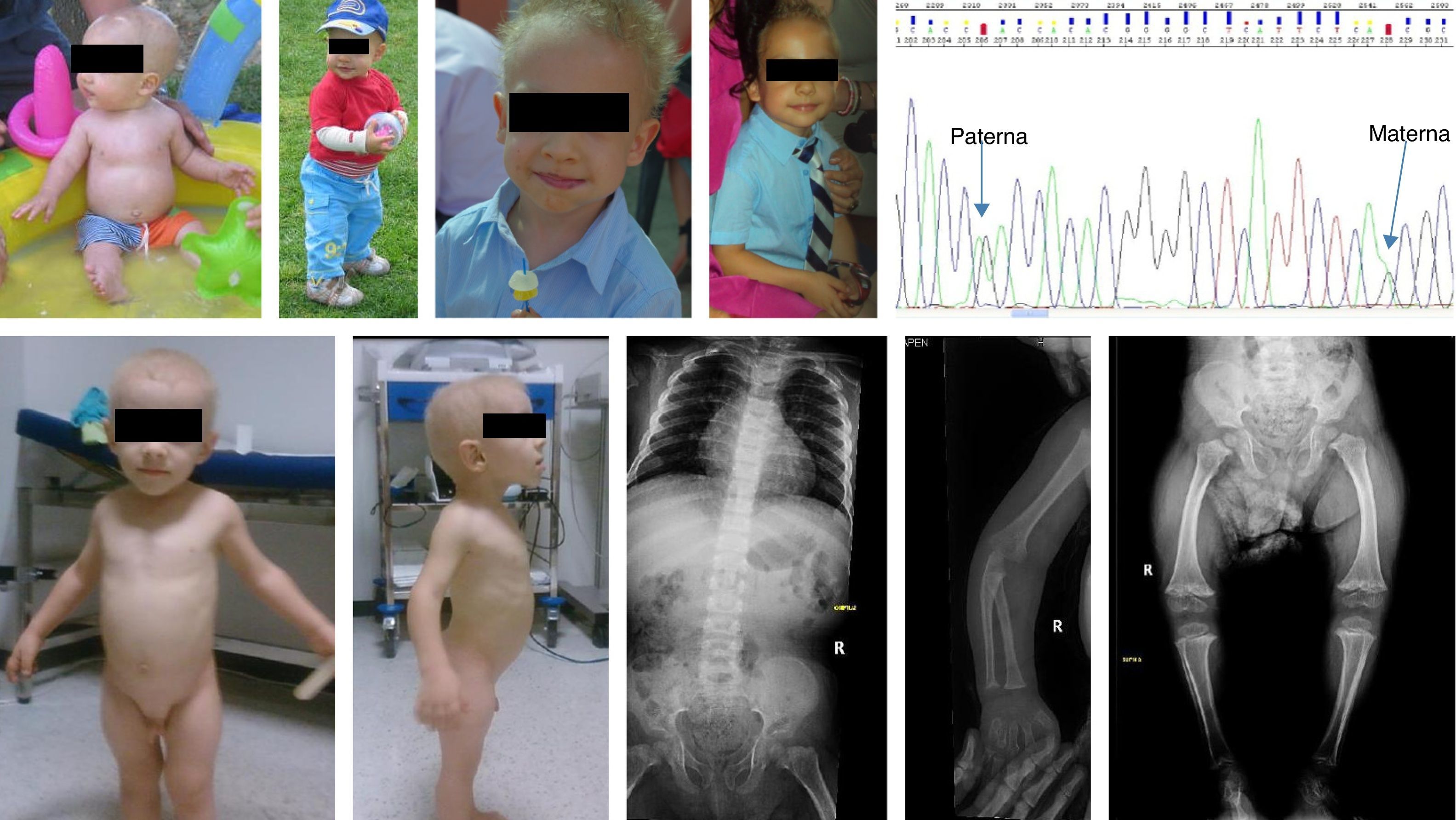
Varón de 2,3 años de origen rumano, con el diagnóstico de acondroplasia desde la semana de vida. Embarazo fruto de padres no consanguíneos controlado en Rumania, parto a término del que se desconocen los datos antropométricos. A la exploración presenta una talla de 62cm (−9,6DE), peso de 8.620g (−3,34DE), acortamiento de extremidades superiores e inferiores, manos toscas y dedos cortos. No deformidad craneal con escasa cantidad de pelo, muy fino y rubio. Estudio óseo: huesos de las manos cortos con metáfisis en copa, rodillas con metáfisis anchas e irregulares y epífisis poco osificadas, incurvación de los fémures con retraso en la osificación de la epífisis femoral proximal, caderas con acetábulos normales (fig. 1). Se realiza estudio analítico (hemograma, bioquímica e inmunoglobulinas) con normalidad de los parámetros estudiados. A los 7,4 años presenta 90,6cm (−6,2DE), velocidad de crecimiento de entre 4 y 7cm al año (−2,1DE y −1,4DE), no ha padecido ninguna infección grave ni cuadro de malabsorción intestinal. Se solicita estudio directo del gen RMRP donde se observan 2 mutaciones en heterocigosis [g.96_97dupTG, materno; g.-25-11 dupACTACTCTGTGAAGC]. El padre no colabora (fig. 1).
Las mutaciones encontradas en estos 2 casos han sido descritas previamente en pacientes con fenotipo de hipoplasia cartílago-pelo de origen europeo4. La mutación g.-25-11 dupACTACTCTGTGAAGC del paciente rumano está descrita en un paciente de origen español5. El diagnóstico diferencial se contempla en la tabla 1. Un diagnóstico completo, tanto clínico como molecular, permite predecir y realizar un mejor seguimiento de la enfermedad. Se ha de estudiar la función inmunológica tanto humoral (déficit de IgG) como celular (linfopenia), así como la presencia de anemia macrocítica no regenerativa. Además, pueden asociar neoplasias, principalmente de origen hematológico (linfomas y leucemia)6. Las opciones terapéuticas contemplan las osteotomías para corregir deformidades, el alargamiento óseo, el tratamiento de las infecciones con profilaxis de las mismas si existe inmunodeficiencia y de la anemia si aparece. El diagnóstico molecular permite un adecuado consejo genético tanto familiar como reproductivo.
Diagnóstico diferencial de la hipoplasia de pelo-cartílago
| Hipoplasia de pelo cartílago | Displasia cifomélica | Displasia inmuno ósea tipo Schimke | Síndrome de Omenn | Displasia esquelética con inmunodeficiencia combinada severa | Síndrome de Shwachman-Diamond | |
|---|---|---|---|---|---|---|
| MIM | 250250 | 211350 | 242900 | 603554 | 200900 | 260400 |
| Gen | RMRP | Desconocido | SMARCAL1 | DCLRE1C,RAG1,RAG2 | Desconocido | SBDS |
| Localización | 9p13 | Desconocido | 2q35 | 11p12,10‘13 | Desconocido | 7q11 |
| Herencia | AR | AR | AR | AR | AR | AR |
| Alt. esq | POS | POS | POS | NEG | POS | POS |
| Tipo | Metafisaria | Metafisaria | Espondiloepifisaria | — | Metafisaria | Metafisaria |
| Fémur y tibia incurvados | POS | POS | NEG | NEG | NEG | NEG |
| Talla baja | POS | POS | POS | NEG | POS | POS |
| Desproporcionada | Desproporcionada | Desproporcionada | Fracaso del desarrollo | Desproporcionada | ||
| Alt. hemato | POS | NEG | POS | POS | POS | POS |
| Tipo | Linfopenia, neutropenia, riesgo malignidad | Linfopenia, neutropenia, trombocitopenia | Eosinofilia, trombocitopenia | Linfopenia | Pancitopenia, riesgo malignidad | |
| Anemia | Frecuente | NEG | POS | POS | NEG | POS |
| Pelo | Ralo, cejas y pestañas escasas y rubias | Normal | Fino | Alopecia | Normal | Normal |
| Alt. inmuno | POS | NEG | POS | POS | POS | NEG |
| Tipo | ↓Cél T y B | ↓Cél T | Linfoadenopatía, nódulos linfáticos desestructurados, ↓Cél B | Agammaglobulinemia, hipoplasia de timo | ||
| Alt. gastro | POS | NEG | NEG | NEG | NEG | POS |
| Tipo | Malabsorción intestinal, enfermedad de Hirschprung, atresia esofágica | Insuficiencia pancreática exocrina | ||||
| Alt. renal | NEG | NEG | POS | NEG | NEG | NEG |
| Tipo | Nefropatía progresiva, rápida y fatal | |||||
| Retraso mental | NEG | NEG | NEG pero descrito | NEG | DESCONOCIDO | POS |
| Esperanza de vida | Adulto | Adulto | Infancia | Infancia | Lactante | Adulto |
Alt.: alteración; AR: autosómica recesiva; NEG: negativo; POS: postivo.
Expresamos nuestro agradecimiento a los técnicos de laboratorio, D. Jesús Gallego Merlo y D. Camilo Vélez Monsalve por su buen hacer diario y su aportación al diagnóstico de estos 2 casos. Agradecemos a los pacientes y sus familiares el permiso dado para publicar las imágenes y datos clínicos con un objetivo exclusivamente científico.
