




The diagnosis of cartilage-hair hypoplasia, also known as McKusick type metaphyseal chondrodysplasia (OMIM 250250), is based on disproportionate short stature, bowed femurs and tibias, short and broad phalanges, loose ligaments with characteristic incomplete extension of the elbows and sparse hair, and was first described in the Amish population of the United States.1 It is caused by mutations in the RMRP gene (OMIM 157660; 9p13),2 a nuclear noncoding gene for an RNA chain that is a subunit of the mitochondrial RNase MRP protein complex, which is involved in ribosomal assembly and cellular cycle regulation. The disorder has a recessive pattern of inheritance, and depending on the severity of the mutation it can have a broad range of clinical manifestations (from mild to severe): metaphyseal dysplasia without hypotrichosis, cartilage-hair dysplasia and anauxetic dysplasia.3
Case 1Boy, 7 months of age, with healthy nonconsanguineous Spanish parents. He was born at 40 weeks, with a weight of 3200g, length of 45cm and head circumference of 36cm. He was admitted at 4 months of age with acute gastroenteritis. The physical examination revealed sparse, fine and brittle hair, shortened limbs, moderate varus bowing of the femurs, and short fingers. His height was 65.5cm (−1.31SD) and his weight 7850g (−0.72SD). Bone radiography showed shortening of long bones and metaphyseal widening. Immunologic testing showed moderate neutropaenia and lymphocytopaenia (reduced T CD4+ and B+T cells); low levels of IgA and normal IgM and IgG levels. These findings led to a clinical diagnosis of cartilage-hair hypoplasia. An RMRP test was ordered at 3 years of age, which detected the presence of 2 heterozygous mutations [g.236A>G, paternal; g.260G>A, maternal]. This diagnosis allowed for screening the maternal blood for the paternal mutation in the foetal DNA (noninvasive prenatal technique) at 8 and 10 weeks in the 2 subsequent gestations of this couple. In the first of these pregnancies, the paternal mutation in the foetal DNA was detected in maternal blood and confirmed by amniocentesis (invasive technique). The pregnancy was terminated. The paternal mutation was not detected in the second pregnancy, and this finding was confirmed by an invasive prenatal test (healthy noncarrier foetus, publication in progress). At 9 years of age, the patient still has short stature (height 108cm [−4.06SD]; weight 20kg [−2.25SD]), and moderate varus bowing of the limbs with followup in the rehabilitation department. His hair is sparse. The patient has had 2 episodes of pneumonia in which no pathogen was identified. His vaccinations are up to date with the immunisation schedule of his autonomous community, and the patient had mild febrile reactions to live virus vaccines. The patient is receiving prophylaxis with trimethoprim-sulfamethoxazole 3 days a week (Fig. 1).
Case 2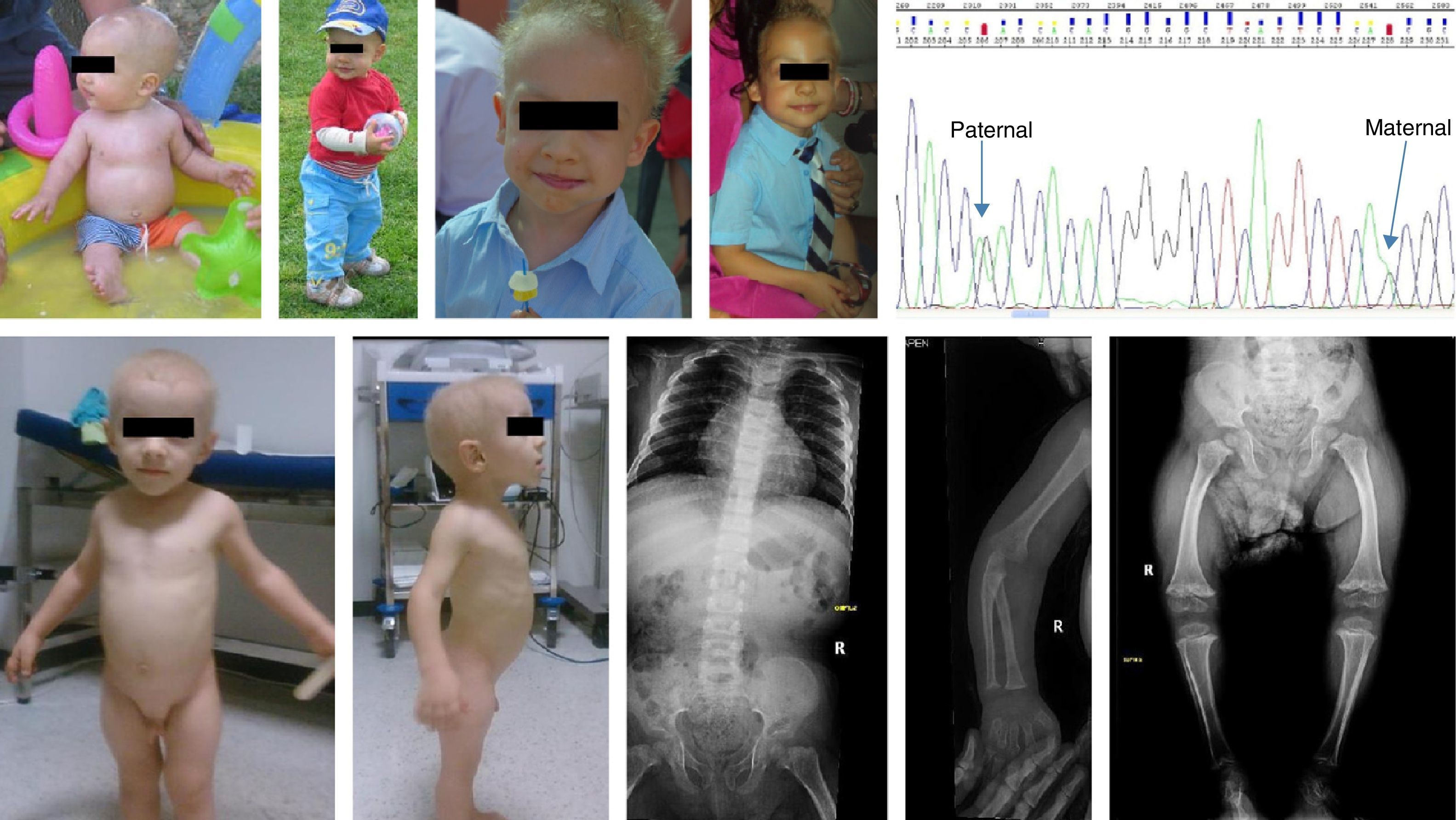
Boy aged 2.3 years of Romanian descent diagnosed with achondroplasia at one week of life. The parents were nonconsanguineous, the pregnancy was monitored in Romania, and the patient was born to term; the birth anthropometric measurements are not known. Physical examination showed a height of 62cm (−9.6SD), weight of 8620g (−3.34SD), shortening of upper and lower extremities, thick hands and short fingers. There were no skull abnormalities, and the hair was sparse, very fine and blonde. Bone radiography showed short hand bones with cone-shaped epiphyses, knees with broad and irregular metaphyses and poorly ossified epiphyses, femoral bowing with delayed ossification of the proximal femoral epiphyses, and hip bones with normal acetabula (Fig. 1). Laboratory tests were performed (complete blood count, biochemistry panel, and immunoglobulins) the results of which were normal. At 7.4 years of age he was 90.6cm tall (−6.2SD), had a growth rate of 4–7cm a year (−2.1SD and −1.4SD), he had not developed any severe infections or any type of gastrointestinal malabsorption. We ordered a direct RMRP gene test that revealed 2 heterozygous mutations (g.96_97dupTG, maternal; g.-25-11 dupACTACTCTGTGAAGC). The father did not cooperate (Fig. 1).
The mutations found in these 2 cases have been described previously in patients of European descent with a cartilage-hair hypoplasia phenotype.4 The g.-25-11 dupACTACTCTGTGAAGC mutation in the Romanian patient has been described in a patient of Spanish descent.5Table 1 shows the differential diagnosis. A full diagnosis based on clinical and molecular findings can help predict the prognosis of the disease and inform its management. Immune function, both humoral (IgG deficiency) and cellular (lymphocytopaenia), and the presence of nonregenerative macrocytic anaemia should be evaluated. These patients may also develop malignancies, especially of the blood (lymphomas and leukaemia).6 Possible treatments include osteostomies to correct deformities, surgical bone lengthening, infection prophylaxis in the presence of an immunodeficiency, and prophylactic treatment for anaemia, should it develop. Molecular diagnosis makes it possible to provide adequate genetic counselling for family planning and reproductive health.
Differential diagnosis of cartilage-hair hypoplasia.
| Cartilage-hair hypoplasia | Kyphomelic dysplasia | Immunoosseous dysplasia, Schimke type | Omenn syndrome | Skeletal dysplasia with severe combined immunodeficiency | Shwachman–Diamond syndrome | |
|---|---|---|---|---|---|---|
| OMIM | 250250 | 211350 | 242900 | 603554 | 200900 | 260400 |
| Gene | RMRP | Unknown | SMARCAL1 | DCLRE1C, RAG1, RAG2 | Unknown | SBDS |
| Location | 9p13 | Unknown | 2q35 | 11p12,10’13 | Unknown | 7q11 |
| Inheritance | AR | AR | AR | AR | AR | AR |
| Skeletal abn. | POS | POS | POS | NEG | POS | POS |
| Type | Metaphyseal | Metaphyseal | Spondyloepiphyseal | – | Metaphyseal | Metaphyseal |
| Bowed femur and tibia | POS | POS | NEG | NEG | NEG | NEG |
| Short stature | POS | POS | POS | NEG | POS | POS |
| Disproportionate | Disproportionate | Disproportionate | Growth failure | Disproportionate | ||
| Haematological abn. | POS | NEG | POS | POS | POS | POS |
| Type | Lymphocytopaenia, neutropaenia, risk of malignancy | Lymphocytopaenia, neutropaenia, thrombocytopaenia | Eosinophilia, thrombocytopaenia | Lymphocytopaenia | Pancytopaenia, risk of malignancy | |
| Anaemia | Frequent | NEG | POS | POS | NEG | POS |
| Hair | Thin, sparse and blonde eye brows and lashes | Normal | Fine | Alopecia | Normal | Normal |
| Immunological abn. | POS | NEG | POS | POS | POS | NEG |
| Type | ↓ T and B cells | ↓ T cells | Lymphadenopathy, architectural effacement of lymph nodes,↓ B cells | Agammaglobulinaemia, thymic hypoplasia | ||
| Gastrointestinal abn. | POS | NEG | NEG | NEG | NEG | POS |
| Type | Gastrointestinal malabsorption, Hirschprung disease, oesophageal atresia | Exocrine pancreatic insufficiency | ||||
| Kidney abn. | NEG | NEG | POS | NEG | NEG | NEG |
| Type | Progressive nephropathy, rapid and fatal | |||||
| Intellectual disability | NEG | NEG | NEG but described | NEG | Unknown | POS |
| Life expectancy | Adulthood | Adulthood | Childhood | Childhood | Infancy | Adulthood |
Abn.: abnormality; AR: autosomal recessive; NEG: negative; POS: positive.
We want to thank the laboratory technicians, Jesús Gallego Merlo and Camilo Vélez Monsalve, for their invaluable daily work and their contribution to the diagnosis of these 2 cases. We thank the patients and their families for consenting to the publications of images and clinical data for scientific purposes.
Please cite this article as: Fenollar-Cortés M, Lara-Orejas E, González-Meneses A, Ruibal-Francisco JL, Trujillo-Tiebas MJ. El diagnóstico clínico-molecular en la hipoplasia de cartílago-pelo: dos nuevos casos. An Pediatr (Barc). 2015;82:436–439.
