

La distrofia miotónica es una enfermedad multisistémica autosómica dominante de expresividad variable. Se revisa nuestra experiencia de 18 años en pacientes afectados.
ResultadosSe han identificado 11 pacientes confirmados con el estudio genético molecular: 2 fallecieron, 5 siguen en control, a 2 se los sigue en otro centro y 3 abandonaron el control. Tres son familiares entre sí. Iniciaron en el período neonatal 7 niños con hipotonía, 4 de ellos con sufrimiento fetal añadido. Un niño se diagnosticó a los 3 meses por el padre afectado. Una niña consultó a los 10 años por agarrotamientos de las manos desde hacía años, un niño consultó a los 5 años con posturas anómalas de las manos y un niño consultó a los 4 años por retraso psicomotor. Alteraciones asociadas: 7 niños con retraso psicomotor, 2 casos de cataratas, un caso de diabetes de tipo 1, 3 casos de hipercolesterolemia, un sarcoma de pared abdominal, un caso de fractura de fémur y cadera, 2 casos de comunicación interauricular. El diagnóstico se realizó en 5 casos por la clínica o el fenotipo de madre y niño, en 3 casos tras diagnóstico familiar y en los 3 casos no congénitos sintomáticos exclusivamente por la clínica del niño.
DiscusiónLa distrofia miotónica es poco frecuente en nuestra experiencia; más frecuentes son las formas congénitas, que asocian con frecuencia sufrimiento perinatal. La genética permite identificar o excluir el proceso. Debe realizarse ante recién nacidos hipotónicos de causa no aclarada y plantearse en niños ante alteraciones motrices en los dedos y las manos no fácilmente explicables.
Myotonic dystrophy is a highly variable autosomic dominant inherited multisystemic disease. We review our 18 years experience with patients suffering from this disease.
ResultsEleven patients were identified following a molecular genetic study: 2 patients died, 5 are still under control, 2 are being controlled in another Centre, and 3 dropped out. Three of them were relatives. Seven newborns started with hypotonic symptoms in the neonatal period, with hypotonic symptoms, of which 4 had foetal suffering. One child was diagnosed at age of 3 due to her father being affected. One girl was seen at age of 10 due to stiffness and tightening of her hands for years. One boy, aged 5, was examined due to abnormal hands posture, and a 4 year old child due to psychomotor delay. Associated disorders: 7 children with psychomotor delay, 2 cases of cataracts, 1 case of diabetes type I, 3 cases of hypercholesterolemia, 1 abdominal sarcoma, 1 case of femur and hip fracture, 2 cases of interatrial communication. The diagnostic was made in 5 cases by a clinic due to mother-son relation phenotype, in 3 cases after the family diagnosis and in another 3 cases non-congenital symptoms exclusively in the child's clinic.
DiscussionIn our experience, myotonic dystrophy is uncommon; it is often congenital, and is associated with perinatal suffering. Genetics can identify or exclude the process. This must be done on newborns who are hypotonic for an unknown reason. It should be suspected in a child who presents with motor abnormalities in the fingers and hands.
La distrofia miotónica, o enfermedad de Steinert, es una enfermedad multisistémica de herencia autosómica dominante con penetrancia casi completa y expresividad variable. Se asocia a deterioro del músculo liso, los sistemas nervioso central y endocrino, el ojo, el hueso, la piel, el aparato respiratorio y los sistemas inmunitario y hematopoyético.
Se debe a una amplificación de un triplete CTG en el extremo 3’ no transcrito de un gen situado en el cromosoma 19q3, codificante para la proteína DMCK de la familia de las proteincinasas. En los individuos normales, el número de tripletes oscila entre 5 y 37. Las formas tardías parciales, dominadas por los síntomas oftalmológicos, presentan entre 50 y 100 repeticiones. Los afectados de la forma congénita tienen hasta 2.500.
La forma clásica se presenta habitualmente a partir de la adolescencia. Hay una combinación variable, con amplios grados de gravedad, de fenómeno miotónico y debilidad amiotrofiante progresiva, que afecta preferentemente a la musculatura facial. Hay también una gran variabilidad en la presentación de síntomas asociados, que incluyen calvicie, alteraciones de la glucorregulación, cataratas, miocardiopatía, atrofia gonadal, afectación de músculo liso, somnolencia y deficiencia mental o demencia.
La distrofia miotónica congénita se transmite casi siempre por vía materna. Se presenta en período neonatal con hipotonía e hipoactividad generalizada, incluida diplejía facial, y alteraciones respiratorias y de succión-deglución. La evolución, si sobreviven a los primeros meses, suele ser de mejoría progresiva, con retraso mental y variable aparición de manifestaciones de la forma clásica. El fenómeno miotónico puede aparecer en la infancia, pero no antes de los 3 o 4 años. Es posible que la madre no presente manifestaciones, ni siquiera desde el punto de vista electromiográfico, en el momento de tener un hijo afectado.
Se revisa nuestra experiencia en pacientes con distrofia miotónica en la Sección de Neuropediatría durante un período de 18 años.
Pacientes y métodosSe han revisado los casos de distrofia miotónica de la base de datos de Neuropediatría del Hospital Universitario Miguel Servet de Zaragoza desde su puesta en funcionamiento en mayo de 1990 a 26 de julio de 2008.
Se revisan, también, los pacientes en los que se realizó estudio genético de distrofia miotónica que excluyó este diagnóstico.
Para el diagnóstico genético se utilizó la técnica de PCR, cebadores específicos y Southern hibridado con la Sonda Sb1.4 (cADN 25) y digestiones con enzimas Eco R1, Bgl 1.
ResultadosEn la base de datos de Neuropediatría del Hospital Universitario Miguel Servet de Zaragoza1,2 en el período de estudio de 18 años están incluidos 10.889 niños. Tienen el diagnóstico de distrofia miotónica 13 casos. Se excluyeron 2 pacientes sobre los que no se había realizado un seguimiento; únicamente habían ingresado en nuestro hospital por otros procesos y se los controlaba en otras comunidades. En la revisión se incluyen, por tanto, 11 pacientes, todos con estudio genético positivo de distrofia miotónica (tabla 1). Cinco siguen en control, 2 fallecieron (las formas neonatales más graves), 2 se controlan en Teruel y 2 abandonaron el control media de seguimiento en consulta de 4,7 años; rango: 21 días-12 años. Para facilitar la exposición hemos asignado un número a cada paciente.
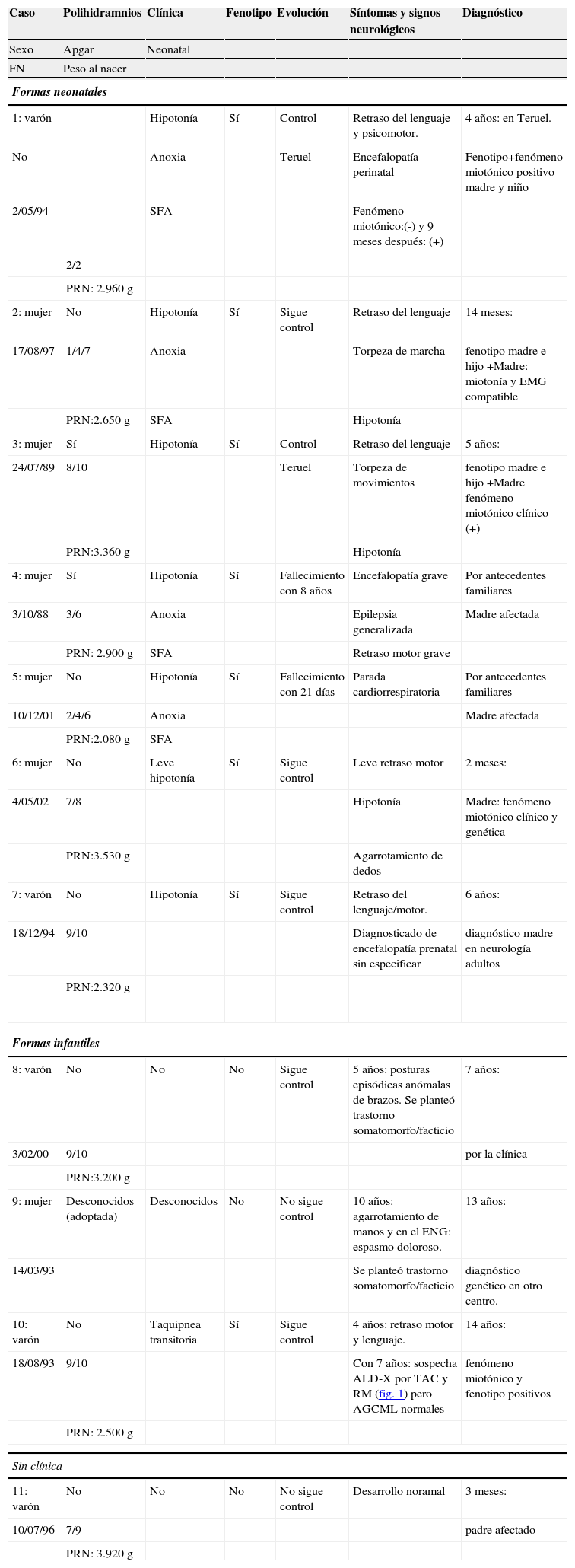
Datos perinatales, neurológicos y diagnósticos
| Caso | Polihidramnios | Clínica | Fenotipo | Evolución | Síntomas y signos neurológicos | Diagnóstico |
| Sexo | Apgar | Neonatal | ||||
| FN | Peso al nacer | |||||
| Formas neonatales | ||||||
| 1: varón | Hipotonía | Sí | Control | Retraso del lenguaje y psicomotor. | 4 años: en Teruel. | |
| No | Anoxia | Teruel | Encefalopatía perinatal | Fenotipo+fenómeno miotónico positivo madre y niño | ||
| 2/05/94 | SFA | Fenómeno miotónico:(-) y 9 meses después: (+) | ||||
| 2/2 | ||||||
| PRN: 2.960g | ||||||
| 2: mujer | No | Hipotonía | Sí | Sigue control | Retraso del lenguaje | 14 meses: |
| 17/08/97 | 1/4/7 | Anoxia | Torpeza de marcha | fenotipo madre e hijo +Madre: miotonía y EMG compatible | ||
| PRN:2.650g | SFA | Hipotonía | ||||
| 3: mujer | Sí | Hipotonía | Sí | Control | Retraso del lenguaje | 5 años: |
| 24/07/89 | 8/10 | Teruel | Torpeza de movimientos | fenotipo madre e hijo +Madre fenómeno miotónico clínico (+) | ||
| PRN:3.360g | Hipotonía | |||||
| 4: mujer | Sí | Hipotonía | Sí | Fallecimiento con 8 años | Encefalopatía grave | Por antecedentes familiares |
| 3/10/88 | 3/6 | Anoxia | Epilepsia generalizada | Madre afectada | ||
| PRN: 2.900g | SFA | Retraso motor grave | ||||
| 5: mujer | No | Hipotonía | Sí | Fallecimiento con 21 días | Parada cardiorrespiratoria | Por antecedentes familiares |
| 10/12/01 | 2/4/6 | Anoxia | Madre afectada | |||
| PRN:2.080g | SFA | |||||
| 6: mujer | No | Leve hipotonía | Sí | Sigue control | Leve retraso motor | 2 meses: |
| 4/05/02 | 7/8 | Hipotonía | Madre: fenómeno miotónico clínico y genética | |||
| PRN:3.530g | Agarrotamiento de dedos | |||||
| 7: varón | No | Hipotonía | Sí | Sigue control | Retraso del lenguaje/motor. | 6 años: |
| 18/12/94 | 9/10 | Diagnosticado de encefalopatía prenatal sin especificar | diagnóstico madre en neurología adultos | |||
| PRN:2.320g | ||||||
| Formas infantiles | ||||||
| 8: varón | No | No | No | Sigue control | 5 años: posturas episódicas anómalas de brazos. Se planteó trastorno somatomorfo/facticio | 7 años: |
| 3/02/00 | 9/10 | por la clínica | ||||
| PRN:3.200g | ||||||
| 9: mujer | Desconocidos (adoptada) | Desconocidos | No | No sigue control | 10 años: agarrotamiento de manos y en el ENG: espasmo doloroso. | 13 años: |
| 14/03/93 | Se planteó trastorno somatomorfo/facticio | diagnóstico genético en otro centro. | ||||
| 10: varón | No | Taquipnea transitoria | Sí | Sigue control | 4 años: retraso motor y lenguaje. | 14 años: |
| 18/08/93 | 9/10 | Con 7 años: sospecha ALD-X por TAC y RM (fig. 1) pero AGCML normales | fenómeno miotónico y fenotipo positivos | |||
| PRN: 2.500g | ||||||
| Sin clínica | ||||||
| 11: varón | No | No | No | No sigue control | Desarrollo noramal | 3 meses: |
| 10/07/96 | 7/9 | padre afectado | ||||
| PRN: 3.920g | ||||||
AGCML: ácidos grasos de cadena muy larga; ALD-X: adrenoleucodistrofia ligada a X; EMG: electromiograma; FN: fecha de nacimiento; SFA: sufrimiento fetal agudo; RM: resonancia magnética; TAC: tomografía axial computarizada.
Los 7 pacientes con forma congénita presentan retraso mental en grado leve-moderado. Los casos 9 y 10 presentan retraso del lenguaje y dificultades de aprendizaje. En los otros 2 casos, la inteligencia es normal y no hay problemas de aprendizaje. Ninguno de los pacientes ha presentado durante el tiempo de seguimiento deterioro muscular ni un fenómeno miotónico significativo con repercusión funcional.
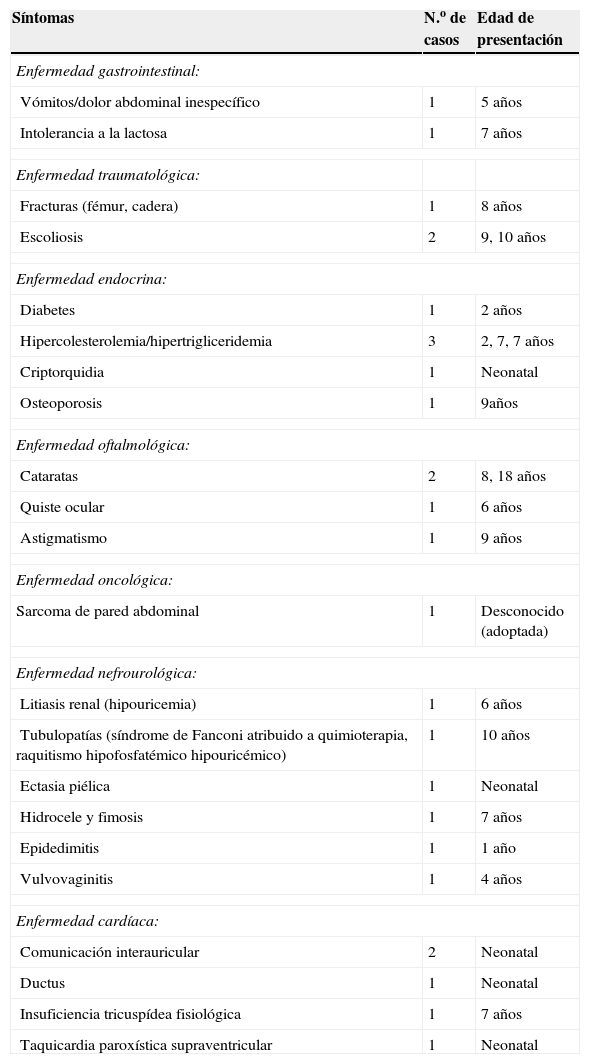
La enfermedad asociada observada se recoge en la tabla 2. La clínica es muy variada.
Problemas no neurológicos asociados
| Síntomas | N.o de casos | Edad de presentación |
| Enfermedad gastrointestinal: | ||
| Vómitos/dolor abdominal inespecífico | 1 | 5 años |
| Intolerancia a la lactosa | 1 | 7 años |
| Enfermedad traumatológica: | ||
| Fracturas (fémur, cadera) | 1 | 8 años |
| Escoliosis | 2 | 9, 10 años |
| Enfermedad endocrina: | ||
| Diabetes | 1 | 2 años |
| Hipercolesterolemia/hipertrigliceridemia | 3 | 2, 7, 7 años |
| Criptorquidia | 1 | Neonatal |
| Osteoporosis | 1 | 9años |
| Enfermedad oftalmológica: | ||
| Cataratas | 2 | 8, 18 años |
| Quiste ocular | 1 | 6 años |
| Astigmatismo | 1 | 9 años |
| Enfermedad oncológica: | ||
| Sarcoma de pared abdominal | 1 | Desconocido (adoptada) |
| Enfermedad nefrourológica: | ||
| Litiasis renal (hipouricemia) | 1 | 6 años |
| Tubulopatías (síndrome de Fanconi atribuido a quimioterapia, raquitismo hipofosfatémico hipouricémico) | 1 | 10 años |
| Ectasia piélica | 1 | Neonatal |
| Hidrocele y fimosis | 1 | 7 años |
| Epidedimitis | 1 | 1 año |
| Vulvovaginitis | 1 | 4 años |
| Enfermedad cardíaca: | ||
| Comunicación interauricular | 2 | Neonatal |
| Ductus | 1 | Neonatal |
| Insuficiencia tricuspídea fisiológica | 1 | 7 años |
| Taquicardia paroxística supraventricular | 1 | Neonatal |
El estudio genético se realizó a 43 pacientes del total de 10.889 niños de la base de datos y resultó negativo en 32 pacientes. El estudio genético se realizó por encefalopatía de presentación neonatal en 5 pacientes, por hipotonía en 15 pacientes, por fenotipo peculiar en 4 pacientes, por retraso del lenguaje, por retraso mental o retraso psicomotor en 7 pacientes y en un paciente por posturas anómalas de manos. El diagnóstico diferencial más frecuente resultante fue el síndrome de Prader-Willi en 5 casos de hipotonía neonatal y la encefalopatía de inicio perinatal sin especificar en 10 casos.
De las 11 familias, se obtuvo el diagnóstico genético del paciente y los familiares en todos los niños, excepto en el caso 9, que no se investigó a la familia por tratarse de una niña adoptada procedente de Costa Rica. En todos ellos el antecedente fue la madre con expansión de la mutación, excepto en el paciente número 11, que fue el caso asintomático en el que ocurrió una regresión de la mutación con respecto a la carga genética paterna.
DiscusiónLa distrofia miotónica es la más frecuente dentro del grupo de distrofias musculares. Se estima una frecuencia de 1/20.000 habitantes; por tanto, no puede considerarse una enfermedad rara3. Sin embargo, es poco frecuente en nuestra experiencia.
En edades pediátricas son más frecuentes las formas neonatales; en nuestra experiencia, 7 casos (el 63,6% de los casos).
Las formas neonatales pueden asociar encefalopatía hipoxicoisquémica, que puede enmascarar el diagnóstico4; hubo sufrimiento fetal agudo en 4 (el 57% del total de nuestros casos de presentación neonatal). Pertenecen a este grupo los 2 fallecimientos de nuestra serie. Las formas neonatales tienen afectación más grave y precoz en relación con el mayor número de repeticiones y con frecuencia, además, como consecuencia de la encefalopatía hipoxicoisquémica asociada.
Se plantea el diagnóstico diferencial con otras causas de hipotonía neonatal, como atrofia muscular espinal, distrofias musculares congénitas y otras miopatías congénitas y diversas encefalopatías, incluidas otras alteraciones cromosómicas5. En la estrategia diagnóstica del recién nacido hipotónico sin orientación clínica clara (como en la atrofia muscular espinal) o por elevación significativa de las enzimas musculares debe considerarse la realización del estudio genético de distrofia miotónica y de enfermedad de Prader-Willi.
Fuera del período neonatal, la estrategia diagnóstica pasa por un elevado índice de sospecha6. Debe plantearse ante retraso psicomotor de causa no aclarada, aunque no existan otros signos, como diplejía facial o el fenómeno miotónico clínico en el paciente (no aparece hasta los 3–4 años) o en su madre. En los 2 pacientes (casos 8 y 9) que iniciaron con posturas anómalas de manos se planteó la posibilidad de un cuadro somatomorfo o facticio. El caso 10 presentaba retraso psicomotor y la exploración por neuroimagen, TC y RM (fig. 1), realizadas a los 8 años, mostraba afectación de sustancia blanca periventricular posterior, que había planteado las posibilidades de leucomalacia periventricular y de adrenoleucodistrofia ligada a X. A los 14 años, en un control en consulta, llamó la atención el fenotipo facial y se comprobó la existencia de fenómeno miotónico clínico positivo. Se han descrito las alteraciones de la RM cerebral en la distrofia miotónica tipo 1. En la forma congénita es característica la dilatación ventricular y moderada o grave afectación de la sustancia blanca posterosuperior a los trígonos, alteraciones no correlacionadas con la edad y, por tanto, debidas fundamentalmente a defecto del desarrollo. En las formas del adulto las alteraciones están relacionadas con la duración de la enfermedad y varían de normalidad a ventriculomegalia y alteraciones de sustancia blanca debidas a degeneración7.

Es importante, con vistas al seguimiento, señalar la gran variedad de enfermedad asociada que se refleja en la tabla 2. Destacan los problemas del lenguaje, los problemas cardíacos8, los problemas endocrinos9,10 como la diabetes y la hipercolesterolemia, y las cataratas, que se presentaron en 2 de nuestros pacientes. Es frecuente, también, la enfermedad gastrointestinal, sobre todo, problemas de deglución y de disfagia11,12, pero también problemas de motilidad intestinal13,14 colelitiasis15 y problemas malabsortivos16,17 a una corta edad.
Para identificar precozmente alteraciones asociadas es aconsejable establecer unos controles periódicos18 con:
- •
Examen clínico con evaluación neurológica anual
- •
Control cardiológico anual
- •
Examen periódico oftalmológico con fondo de ojo y lámpara de hendidura
- •
Control de rehabilitación/ortopedia
- •
Control endocrino periódico: evaluación de función tiroidea, función suprarrenal, pubertad tardía, alopecia, atrofia testicular y diabetes.
- •
Analítica básica con IgG y niveles de colesterol
Existe cierta asociación genotipo/fenotipo, pero resulta arriesgado establecer correlación entre la expansión detectada en el estudio genético y la precocidad en la aparición de síntomas y la gravedad en la evolución del niño. Es reconocido para esta enfermedad el curioso fenómeno de la anticipación, es decir, el aumento de tripletes en generaciones sucesivas de la familia. En la población normal existe estabilidad en el número de repeticiones trasmitidas a la descendencia. Por encima de 37 repeticiones se convierte en inestable, y, además, hay diferencias según sea el origen parental. Además, en las transmisiones maternas existe una tendencia a aumentar la expansión del número de tripletes CTG que puede dar lugar a la forma congénita, letal en el 16% de los casos, o a morbimortalidad en la edad adulta temprana19.
Cuando la transmisión es paterna, aunque se observa el fenómeno de anticipación, en la mayoría de las ocasiones se trasmite, por causas desconocidas, el mismo número de tripletes a la descendencia, pero, en algunos casos, se observa una regresión, o contracción del ADN a partir de un cierto umbral de expansión, posiblemente por un efecto barrera en la línea germinal de individuos masculinos afectados que impide la amplificación de la expansión en los gametos a partir de un cierto grado de expansión parental19. Nuestro caso 11, transmitido por el padre, mostró una regresión de la mutación.
Por último, hay que tener en cuenta la inestabilidad somática, es decir, que el tamaño de la expansión no es la misma en todos los tejidos del individuo.
Es importante el diagnóstico precoz de la distrofia miotónica para evitar incertidumbres y estudios innecesarios, establecer el adecuado seguimiento y, como en toda enfermedad autosómica dominante, establecer el adecuado asesoramiento genético. Para esto es útil establecer estrategias de identificación. No siempre se demuestra el fenómeno miotónico clínico o neurofisiológico en los niños o en sus madres afectadas. Dada la disponibilidad actual del estudio genético, que establece o excluye el diagnóstico, éste se debe realizar ante un recién nacido hipotónico sin diagnóstico establecido y debe plantearse ante retraso psicomotor de causa no aclarada y niños con agarrotamientos o posturas anómalas de manos.
Conflictos de interesesLos autores declaran no tener ningún conflicto de intereses.
