
La discinesia ciliar primaria (DCP) y el síndrome cardioespondilocarpofacial (SCECF) pertenecen al grupo de enfermedades raras, dada su muy baja prevalencia en la población (1/7.554 para la DCP y <1/1.000.000 para el SCECF). Presentamos el caso de una niña con diagnóstico genético inicial de SCECF que, por la mala evolución respiratoria y la presencia de bronquiectasias, se diagnosticó posteriormente de DCP.
Niña con edad actual de 3 años que al nacimiento presentó fenotipo peculiar (frente abombada, filtrum plano, orejas de implantación discretamente bajas, cuello ancho) y síntomas de hipotonía generalizada, dificultades de alimentación, taquipnea, rinitis crónica y acidosis respiratoria. Permaneció ingresada los 3 primeros meses de vida, durante los cuales presentó una atelectasia parcial de lóbulo superior derecho en la radiografía de tórax y una estenosis del agujero magno con compresión de la unión bulbomedular en la resonancia magnética cerebral, por lo que precisó de intervención quirúrgica a los 2 meses de vida.
Se amplió el estudio en este periodo con ecocardiografía, en la que se detectó insuficiencia tricúspidea leve, y un análisis genético de tipo exoma trío dirigido a la sintomatología neurológica, que fue informado con presencia de una variante de novo probablemente patogénica (c.821G>A) en el gen MAP3K7 asociada al SCECF y que engloba retraso del crecimiento, dismorfias faciales, hipotonía, dificultades de alimentación, enfermedad cardíaca y de vértebras cervicales1.
Durante el primer año de vida presentó sintomatología respiratoria crónica consistente en tos húmeda diaria y rinitis, con infecciones respiratorias recurrentes en forma de bronquitis, catarros y otitis serosa crónica bilateral. A la exploración física destacaba la auscultación pulmonar patológica de forma persistente con crepitantes húmedos bilaterales, tiraje intercostal y taquipnea, con saturación de oxígeno normal. Asociaba disfagia orofaríngea diagnosticada por videofluoroscopia con alteración en la eficacia de la deglución y retraso ponderal (percentil 2), por lo que precisó de alimentación por sonda nasogástrica hasta los 5 meses de vida.
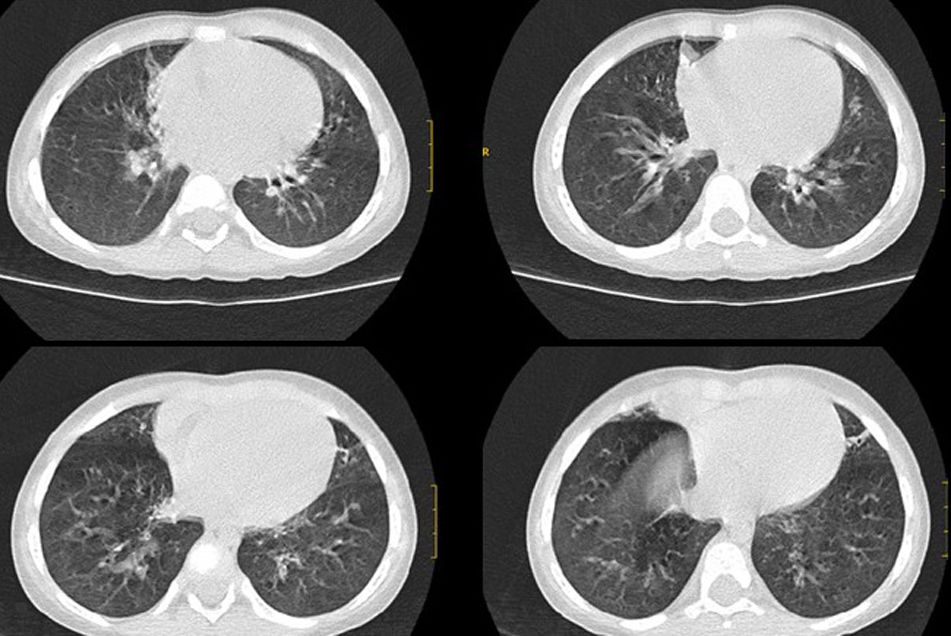
Ante la sospecha de síndrome aspirativo crónico por disfagia y posible reflujo gastroesofágico asociado (descartado posteriormente con pHmetría), se realizó tomografía computarizada pulmonar en la que se apreciaron atelectasias subsegmentarias en el lóbulo medio y la língula con pequeñas bronquiectasias y discreto engrosamiento intersticial peribroncovascular de predominio en lóbulos inferiores (fig. 1). Ante dichos hallazgos se realizó broncoscopia flexible en la que se observó abundante mucosidad en todo el árbol bronquial, con lavado broncoalveolar positivo a Haemophilus influenzae. Como tratamiento recibió budesonida inhalada y nasal, suero salino hipertónico nebulizado, fisioterapia respiratoria y azitromicina oral 3 días por semana.

Dada la mala evolución respiratoria y las escasas complicaciones respiratorias asociadas al SCECF, se plantearon diagnósticos adicionales que cursan con bronquiectasias, como la fibrosis quística o la DCP. A pesar de cribado neonatal con tripsina inmunorreactiva normal, se hizo la prueba del sudor, que también fue normal. Se reanalizó el estudio genético de exoma trío y se informó de la presencia en homocigosis de la mutación c.246+1G>C en el gen ODAD4 o TTC25 asociado a DCP, que codifica el brazo externo de la proteína dineína y ocasiona una disminución grave de la motilidad ciliar2.
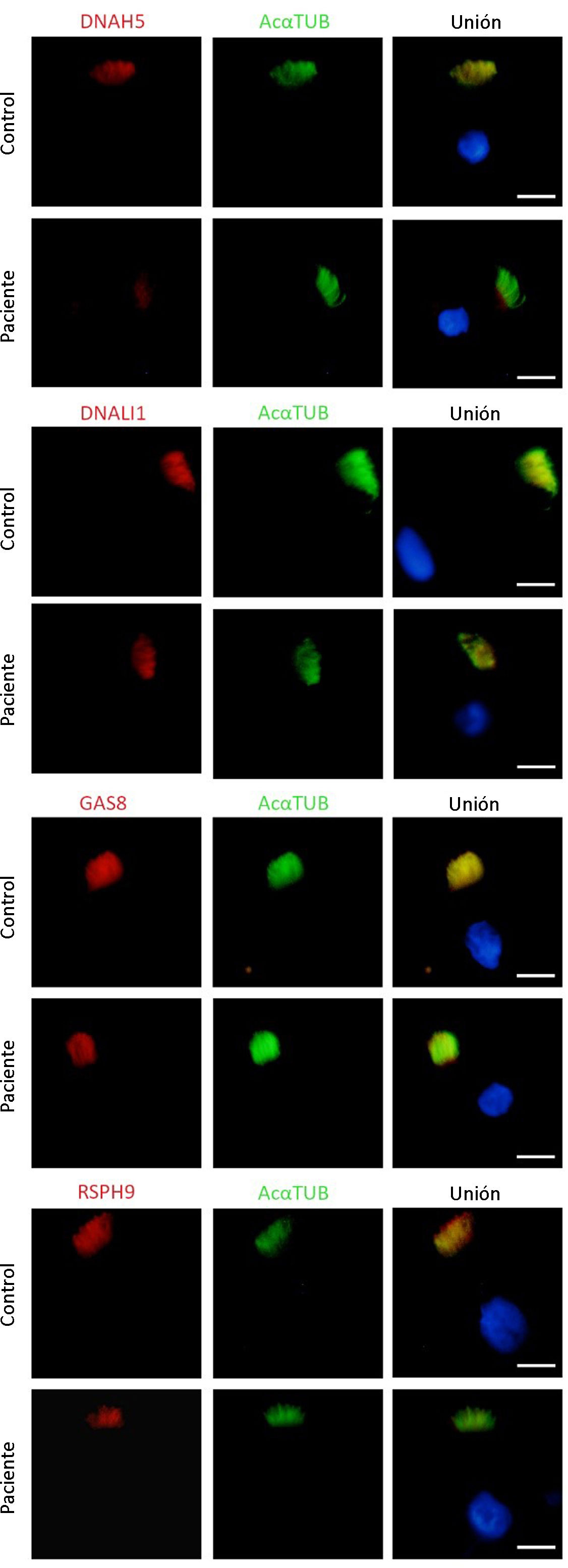
La niña fue derivada al centro de referencia de DCP en el que confirmaron la ausencia de motilidad ciliar (cilios estáticos) mediante videomicroscopia óptica, un resultado bajo de óxido nítrico nasal (ONn) de 13,2 nl/min medido a volumen corriente y una inmunofluorescencia en células ciliadas con el marcador DNAH5 ausente en la mayoría de las células analizadas (fig. 2).
, y α-tubulina acetilada (AcαTUB, en verde). La tercera columna muestra la imagen conjunta con el núcleo marcado con DAPI (en azul). DNAH5 está ausente en el axonema ciliar de la muestra de la paciente comparando con el control. Escala bar, 5μm.")
Análisis de inmunofluorescencia de las proteínas del axonema ciliar en una muestra de epitelio ciliado respiratorio de la paciente y en una muestra control. Localización subcelular de proteínas en el axonema ciliar DNAH5, DNALI1, GAS8 y RSPH9 (en rojo), y α-tubulina acetilada (AcαTUB, en verde). La tercera columna muestra la imagen conjunta con el núcleo marcado con DAPI (en azul). DNAH5 está ausente en el axonema ciliar de la muestra de la paciente comparando con el control. Escala bar, 5μm.
La DCP consiste en un defecto estructural de las células ciliadas, situadas en el tejido respiratorio y gonadal, que altera la motilidad ciliar y disminuye el aclaramiento mucociliar, lo que aumenta el riesgo de infecciones respiratorias. Es un defecto de herencia autosómica recesiva en la mayoría de los casos, con más de 50 mutaciones descritas3 y que asocia síntomas respiratorios, otorrinolaringológicos, esterilidad, embarazo ectópico y, en el 50%, situs inversus con dextrocardia. Desde el punto de vista radiológico presenta engrosamientos peribronquiales con atelectasias o bronquiectasias4.
El diagnóstico se basa en el cribado con el ONn (generalmente disminuido) y se confirma mediante microscopia electrónica (alteraciones ultraestructurales), inmunofluorescencia, videomicroscopia óptica (frecuencia y batido ciliar) y genética5. El tratamiento se basa en la mejora del aclaramiento mucociliar mediante fisioterapia respiratoria, ejercicio aeróbico y agentes mucolíticos (suero salino hipertónico nebulizado); en el control de las infecciones respiratorias (antibioterapia oral o intravenosa en exacerbaciones respiratorias y antibióticos nebulizados) y en terapias antiinflamatorias, como la azitromicina oral6.
Como conclusiones, los autores queremos destacar la importancia de descartar la existencia de otras enfermedades adicionales en todo paciente con un diagnóstico sindrómico confirmado que, por su evolución o sintomatología, no encaje en el fenotipo clínico típico de esa enfermedad, tal y como pasó en nuestra paciente.
Colaboración de los autoresTodos los autores han participado en la elaboración del manuscrito.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses en la elaboración de este manuscrito.

