


El pseudohipoparatiroidismo (PHP) comprende un grupo heterogéneo de enfermedades endocrinológicas que se caracterizan por la existencia de hipocalcemia, hiperfosfatemia y resistencia tisular a la hormona paratiroidea. Se distinguen diferentes formas de PHP. El PHP-Ia es la forma más frecuente y asocia resistencia hormonal múltiple, signos clínicos de osteodistrofia hereditaria de Albright (OHA) y mutaciones en el gen GNAS codificador de la proteína Gsα. El pseudoPHP (PPHP) asocia igualmente mutaciones en el gen GNAS pero cursa con OHA aislada sin anomalías endocrinas. Se presenta una familia con madre afecta de PPHP y dos hijas con PHP-Ia que comparten la misma mutación inactivadora en heterocigosis en el gen GNAS (Asn264LysfsX35). Se discute la diferente expresividad clínica así como el modelo de herencia dominante con impronta genética en el que el fenotipo de la descendencia está deteminado por el sexo del progenitor afecto.
Pseudohypoparathyroidism (PHP) is a heterogeneous group of endocrine diseases characterised by hypocalcaemia, hyperphosphataemia and resistance to PTH. There are different forms of PHP. PHP-Ia is the most frequent form and shows multi-hormonal resistance, GNAS (Gsα) mutations and signs of Albright¿s hereditary osteodystrophy (AHO). PseudoPHP (PPHP) have isolated AHO without hormonal resistance and it is also caused by GNAS mutations. We present a family that share the same inactivating GNAS mutation (Asn264LysfsX35); the mother being affected with PPHP and the two daughters with PHP-Ia. We discuss the different clinical phenotypes and the dominant mode of inheritance with genetic imprinting where the phenotype of the offspring depends on the sex of the parent affected.
El pseudohipoparatiroidismo (PHP) comprende un grupo heterogéneo de enfermedades endocrinológicas que se caracterizan por hipocalcemia, hiperfosfatemia y resistencia a la hormona paratiroidea (PTH). La resistencia parece estar limitada al túbulo renal proximal mientras que las acciones de la PTH se mantienen intactas en otros tejidos1. El mecanismo de acción de la PTH se basa en su unión a un receptor de membrana acoplado a la proteína Gsα (codificada por el gen GNAS), encargada de transmitir la señal biológica a nivel intracelular. Se distinguen dos formas clínicas de PHP en función de la respuesta del AMPc urinario y de la fosfaturia a la admistración exógena de PTH: el tipo I sin respuesta y el tipo II con niveles de AMPc urinario que aumentan pero no la fosfaturia. El PHP-I se clasifica en tres subtipos en función de la presencia de anomalías endocrinas, mutaciones en el gen GNAS (Gsα y presencia o no de Osteodistrofia Hereditaria de Albright (OHA). El PHP-Ia es el más frecuente y asocia mutaciones en el gen GNAS codificador de Gsα, resistencia multihormonal y OHA2,3. El PPHP presenta OHA sin resistencia hormonal. La impronta genética se refiere a la expresión exclusiva o preferencial de un solo alelo según sea el progenitor de origen; se habla de imprinting materno o paterno cuando solamente se expresa el alelo paterno o materno, respectivamente. La impronta se asocia a modificaciones epigenéticas del ADN que ocurren durante la gametogénesis y desarrollo embrionario. Esta expresión genética diferencial puede durar toda la vida o un estadío del desarrollo y ser generalizada o limitada a un tejido. Clínicamente, los posibles efectos de la impronta son que mutaciones con transmisión vertical puedan no manifestarse en generaciones siguientes con aparente falta de penetrancia; la existencia de cuadros clínicos diferentes en caso de deleciones cromosómicas idénticas; la aparición de un fenotipo patológico en caso de disomías uniparentales y el efecto diferencial de las mutaciones somáticas, como ocurre en las neoplasias4.
El locus GNAS (20q13.2-13.3) y los diferentes transcritos para los que codifica (proteínas Gsα, XLαs, NESP55 y transcrito A/B) están sometidos a imprinting. El transcrito Gsα tiene imprinting paterno (sólo es funcional el alelo materno) en hipófisis, tiroides, gónadas y túbulo renal, siendo de expresión bialélica en el resto de tejidos. Se han encontrado mutaciones en el gen GNAS tanto en pacientes con PHP-Ia como en el PPHP5. Se presenta una familia con PHP y una misma mutación en el gen GNAS con fenotipos diferentes reflejando la variabilidad y heterogeneidad genética de esta entidad.
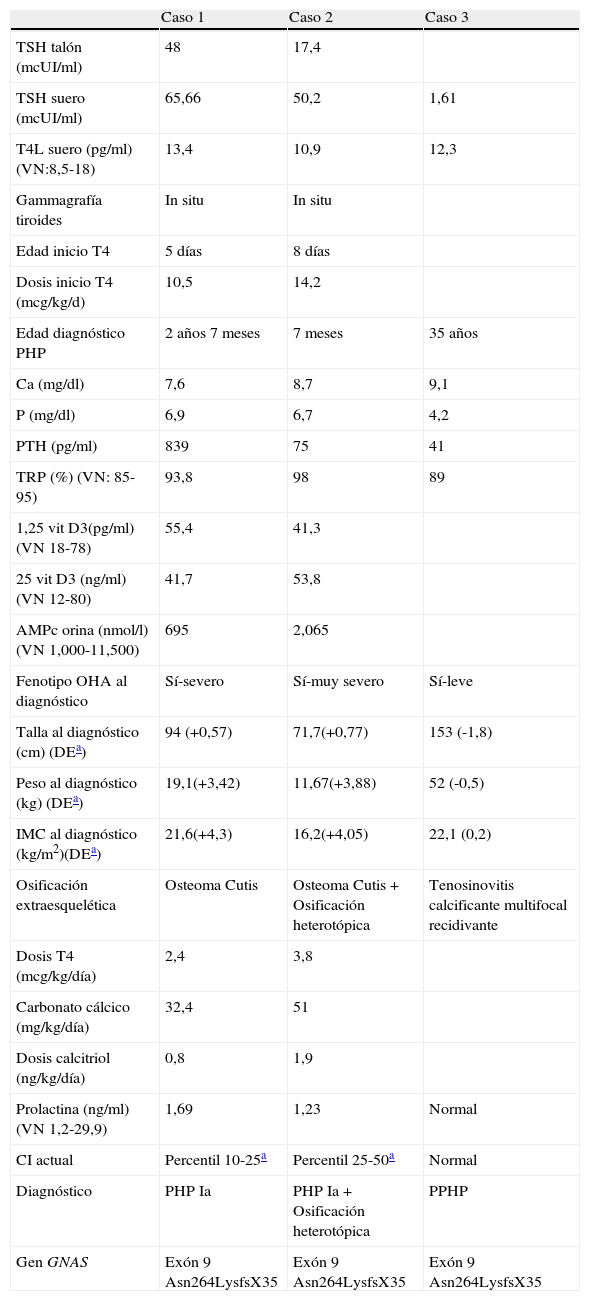
Casos clínicosCaso 1Recién nacida diagnosticada de hipotiroidismo congénito. Dibetes gestacional en embarazo. Padre con hipotiroidismo congénito en tratamiento y fenotipo normal. Hábito tosco, cutis marmorata; ganancia de peso excesiva en primer año de vida y desarrollo motor retardado con desarrollo cognitivo normal. A los 18 meses lesiones máculo-papulosas induradas en tronco y extremidades diagnosticándose por biopsia de osteoma cutis (OC) (fig. 1). Se realiza estudio fosfo-cálcico y se diagnostica de PHP-Ia, iniciándose tratamiento a los 2,7 años. Crecimiento normal con talla y peso en percentil 50. Última densitometría ósea Z-score +2,41 DE.
Caso 2.")
Recién nacida, hermana del anterior caso, diagnosticada de hipotiroidismo congénito. En exploración neonatal se observa fenotipo tosco, discreta macroglosia, hábito abotargado y a los 15 días de vida zona empastada en región mandibular izquierda. A los 4 meses se realiza biopsia objetivándose tejido fibroadiposo que engloba estructuras calcificadas y osificadas. Desarrollo motor lento con desarrollo cognitivo dentro de la normalidad. Lesiones induradas superficiales en tronco y extremidades. Se diagnóstica de PHP-Ia y se inicia tratamiento a los 11 meses. El TAC muestra una masa extensa de calcinosis subcutánea en región mandibular izquierda desde el mentón hasta la articulación temporomandibular izquierda, infiltra componentes blandos articulares y tendinosos profundos y produce una anquilosis de la articulación (osificación heterotópica profunda) (fig. 2). Con 4 años se realiza osteoartrectomía radical y artroplastia reconstructora.
Caso 3.")
Madre intervenida de tenosinovitis calcificante recurrente del nervio mediano y de rodilla. Facies tosca, cutis marmorata, talla 153cm (-1,8 DE), peso 52kg (-0,5 DE), metabolismo calcio-fósforo normal, diagnosticándose de PPHP. No asocia enfermedades concomitantes, presentando desarrollo neurocognitivo dentro de la normalidad.
Estudio genético familiar: mutación en heterocigosis en el exón 9 del gen GNAS (inserción de una adenina), que conlleva la alteración de la pauta de lectura a partir del aminoácido 264 y la generación de una proteína truncada (Asn264LysfsX35) (tabla 1).
Casos clínicos
| Caso 1 | Caso 2 | Caso 3 | |
| TSH talón (mcUI/ml) | 48 | 17,4 | |
| TSH suero (mcUI/ml) | 65,66 | 50,2 | 1,61 |
| T4L suero (pg/ml) (VN:8,5-18) | 13,4 | 10,9 | 12,3 |
| Gammagrafía tiroides | In situ | In situ | |
| Edad inicio T4 | 5 días | 8 días | |
| Dosis inicio T4 (mcg/kg/d) | 10,5 | 14,2 | |
| Edad diagnóstico PHP | 2 años 7 meses | 7 meses | 35 años |
| Ca (mg/dl) | 7,6 | 8,7 | 9,1 |
| P (mg/dl) | 6,9 | 6,7 | 4,2 |
| PTH (pg/ml) | 839 | 75 | 41 |
| TRP (%) (VN: 85-95) | 93,8 | 98 | 89 |
| 1,25 vit D3(pg/ml) (VN 18-78) | 55,4 | 41,3 | |
| 25 vit D3 (ng/ml) (VN 12-80) | 41,7 | 53,8 | |
| AMPc orina (nmol/l) (VN 1,000-11,500) | 695 | 2,065 | |
| Fenotipo OHA al diagnóstico | Sí-severo | Sí-muy severo | Sí-leve |
| Talla al diagnóstico (cm) (DEa) | 94 (+0,57) | 71,7(+0,77) | 153 (-1,8) |
| Peso al diagnóstico (kg) (DEa) | 19,1(+3,42) | 11,67(+3,88) | 52 (-0,5) |
| IMC al diagnóstico (kg/m2)(DEa) | 21,6(+4,3) | 16,2(+4,05) | 22,1 (0,2) |
| Osificación extraesquelética | Osteoma Cutis | Osteoma Cutis+Osificación heterotópica | Tenosinovitis calcificante multifocal recidivante |
| Dosis T4 (mcg/kg/día) | 2,4 | 3,8 | |
| Carbonato cálcico (mg/kg/día) | 32,4 | 51 | |
| Dosis calcitriol (ng/kg/día) | 0,8 | 1,9 | |
| Prolactina (ng/ml) (VN 1,2-29,9) | 1,69 | 1,23 | Normal |
| CI actual | Percentil 10-25a | Percentil 25-50a | Normal |
| Diagnóstico | PHP Ia | PHP Ia+Osificación heterotópica | PPHP |
| Gen GNAS | Exón 9 Asn264LysfsX35 | Exón 9 Asn264LysfsX35 | Exón 9 Asn264LysfsX35 |
Tanto el PHP-Ia como el PPHP asocian mutaciones en heterocigosis que afectan a los exones codificantes de la proteína Gsα del gen GNAS que condicionan una pérdida de función de la proteína. El PHP sigue un modelo de herencia dominante pero la misma mutación se asocia con fenotipos diferentes (PHP-Ia vs PPHP) debido al fenómeno de imprinting, de modo que el fenotipo está determinado por el sexo del progenitor que transmite la mutación. En la familia que se presenta, la madre tiene un PPHP y transmite a sus hijas la mutación pero al ocurrir la mutación en el alelo materno, alelo funcional, se desarrolla un cuadro de PHP-Ia. Sin embargo, cuando la mutación se hereda del padre se desarrolla un PPHP, independientemente de que el padre tenga un PHP-Ia o PPHP. La resistencia a la PTH se desarrolla sólo cuando la mutación se hereda de la madre, mientras que el fenotipo OHA se desarrolla cuando se hereda de cualquiera de los dos progenitores. Esta expresión diferencial se explica porque en el túbulo renal, tiroides, hipófisis y gónadas, el transcrito Gsα está sometido a imprinting paterno y si la mutación está presente en el alelo materno, el activo, la funcionalidad de la proteína quedaría a expensas del alelo paterno, pero como está improntado no se expresa, con una ausencia total de la proteína y un cuadro de resistencia a las hormonas que actúan a través de Gsα6,7. El estudio familiar que se presenta es concordante con este modelo de herencia dominante con fenómeno de imprinting.
La misma mutación de GNAS puede presentarse con una severidad variable y diversidad fenotípica tanto para casos esporádicos como para casos intrafamiliares, como así lo demuestra la familia que se presenta8,9. Esta variabilidad se pueden explicar por modificaciones epigenéticas no conocidas y/o por la compleja regulación transcripciónal del gen GNAS o por la participación de genes modificadores diferentes de GNAS o por factores ambientales10–12. Esta heterogeneidad genética se manifiesta en la presencia de fenotipos distintos (PPHP vs PHP-Ia) y en que el grado de severidad de la resistencia hormonal (PHP-Ia) varía entre cada paciente, incluso dentro de la misma hermandad, como ocurre en nuestro caso. El caso 2 es más severo (comienzo más temprano, lesiones de OC más extensas, calcificación profunda en hemicara izquierda, requerimiento de más dosis de tiroxina, carbonato cálcico y de calcitriol), indicando que otros factores genéticos influyen en la expresividad del cuadro. Este caso presenta una osificación heterotópica profunda, típica de la heteroplasia ósea progresiva (HOP), caracterizada por una osificación heterotópica severa y progresiva que afecta a músculos y tejidos conectivos profundos en ausencia de OHA y resistencia hormonal y se asocia también con mutaciones inactivadoras en heterocigosis en el gen GNAS13,14. El fenotipo HOP solamente aparece cuando la mutación es heredada del padre, pero no de la madre15. Algunas mutaciones identificadas en la HOP son idénticas a otras halladas en el PHP-Ia y en el PPHP sugiriendo que las mutaciones del gen GNAS no son el único elemento fisopatológico implicado. No es frecuente que la HOP y PHP-Ia ocurran en el mismo paciente ya que se asocian con una transmisión alelo-específica diferente (padre vs madre), pero se han descrito casos aislados que comparten manifestaciones de las dos entidades16,17. Posiblemente, la HOP represente el extremo más severo de las mutaciones inactivadoras del gen GNAS que cursan con osificación extraesquelética. El mayor grado de osificación heterotópica en algunos pacientes con PHP-Ia se podría explicar por el desarrollo de una segunda mutación somática en el alelo sano, en este caso en el alelo paterno. Esta teoría se apoya en que las osificaciones ectópicas en el PPHP, PHP-Ia y HOP siguen una distribución en mosaico si bien esta teoría no ha sido demostrada.
Gsα es una proteína de señalización intracelular de diferentes hormonas, factores de crecimiento y neurotransmisores. Sin embargo, en el PHP-Ia solamente se afectan una serie de ejes hormonales como TSH, GHRH, LH/FSH o déficits olfatorios y de prolactina, pero no otros como glucagón, vasopresina, CRH, ACTH, isoproterenol, debido a la expresión monoalélica de Gsα solamente en algunos tejidos18. Es posible también que el grado de imprinting paterno de Gsα no sea igual ni completo en todos los tejidos, contribuyendo a explicar la variabilidad clínica a nivel tiroideo, gonadal e hipofisario de los pacientes con PHP-Ia. Estos pacientes presentan un moderado retraso intelectual de patogenia no aclarada y que no parece depender del hipotiroidismo congénito sino de las acciones neuronales de la proteína Gsα.
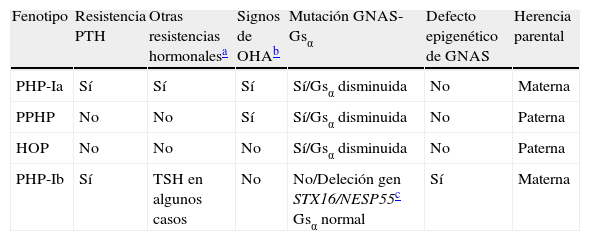
El PHP-Ib se caracteriza por ausencia de OHA y presentar resistencia renal a la PTH y en ocasiones parcial a la TSH con actividad normal de la proteína Gsα. Estudios recientes asocian la presencia de PHP-Ib con defectos en genes reguladores de la metilación del gen GNAS materno (STX16, NESP55) que determinan una alteración de la impronta de los transcritos del locus GNAS diferentes de la proteína Gsα. Se han descrito formas leves de PHP-Ia en los que no se han encontrado mutaciones en los exones que codifican Gsα que presentan defectos de metilación semejantes a los encontrados en el PHP-Ib indicando un cierto solapamiento entre estas dos entidades, pensándose que todos los pacientes con resistencia a la PTH y signos de OHA deberían ser estudiados a nivel molecular no sólo para el gen Gsα sino también para los defectos epigenéticos de GNAS19,20 (tabla 2).
Características clínicas y moleculares asociados con diferentes formas de PHP-I
| Fenotipo | Resistencia PTH | Otras resistencias hormonalesa | Signos de OHAb | Mutación GNAS- Gsα | Defecto epigenético de GNAS | Herencia parental |
| PHP-Ia | Sí | Sí | Sí | Sí/Gsα disminuida | No | Materna |
| PPHP | No | No | Sí | Sí/Gsα disminuida | No | Paterna |
| HOP | No | No | No | Sí/Gsα disminuida | No | Paterna |
| PHP-Ib | Sí | TSH en algunos casos | No | No/Deleción gen STX16/NESP55c Gsα normal | Sí | Materna |
La diferente expresividad clínica y la heterogeneidad genética son dos características del PHP ligado a mutaciones del gen GNAS siendo necesaria la identificación de la mutación en los casos de PPHP y PHP-Ia para poder realizar un consejo genético adecuado.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
