





El síndrome lácrimo-aurículo-dento-digital (síndrome LADD o también conocido como síndrome de Levy-Hollister) es un defecto autosómico dominante con expresividad variable. Este síndrome se caracteriza por la asociación de displasias en diversos órganos y sistemas que afectan a las estructuras craneofaciales, incluyendo glándulas lagrimales y salivales, dientes, oído interno y externo y el esqueleto óseo.
Presentamos este síndrome inusual y desconocido (es el primer caso en nuestro estado del que existen solamente cerca de 100 casos descritos, distribuidos en pocas familias) en referencia a un adolescente de 17 años ingresado en la Unidad de Cuidados Intensivos Pediátricos por otro motivo y que tras la exploración física se llega a un diagnóstico clínico en toda la rama familiar.
Lacrimo-auriculo-dento-digital syndrome (LADD syndrome), also known as Levy-Hollister syndrome, is an autosomal dominant defect with variability on phenotypical expression. This syndrome is characterised by the association of dysplasia in various organs and systems that affect craniofacial structures, including lachrymal and salivary glands, teeth, internal and external ear, and the bone skeleton.
We present this unusual and almost unknown syndrome (the first case in our state, with only about 100 cases described in the world, distributed in a few families) in a teenager of 17 years admitted to PICU for another reason. After the physical examination, a clinical diagnosis was made in the entire family branch.
El síndrome lácrimo-aurículo-dento-digital (síndrome LADD), también conocido como síndrome de Levy-Hollister (OMIM 149730), se caracteriza por asociar alteraciones lacrimales (agenesia de conductos lacrimales), auriculares (pabellones de implantación baja y en forma de copa, sordera neurosensorial o de transmisión), dentales (displasias del esmalte, microdontia, hipodoncia) y esqueléticas distales (clinodactlia, sindactilia, ectrodactilia en manos o pies1-4).
Se trata de una anomalía genética autosómica dominante con una expresividad variable y en ocasiones casos de novo. Es una entidad poco conocida, pues hasta ahora solo se han descrito pocos casos en la literatura1-5.
Dado que este síndrome presenta diferentes niveles de expresión (incluso dentro de una misma rama familiar), con unas alteraciones dismorfológicas características, destacamos la importancia de su diagnóstico clínico.
Caso clínicoAdolescente de 17 años, ingresado en la Unidad de Cuidados Intensivos Pediátricos (UCIP) por embolismo graso secundario a fractura diafisaria de fémur intervenida tras accidente de tráfico. A las 24 h del accidente presentó una disminución del nivel de consciencia, por lo que tuvo que ser intubado y conectado a ventilación mecánica. A la exploración física durante el ingreso destaca también ectrodactilia en extremidades, alteraciones dentarias y auriculares. A los 3 días aparecieron en el tórax y las conjuntivas lesiones petequiales. Se practicaron una tomografía computarizada y una resonancia magnética craneales, que mostraron lesiones frontales hipóxico isquémicas, compatibles con síndrome de embolismo graso. La evolución fue favorable.
La presencia de varias alteraciones a nivel de extremidades, dientes y pabellones auriculares nos orientó a la presencia de un síndrome con entidad propia por lo que se realizó un estudio clínico dismorfológico.
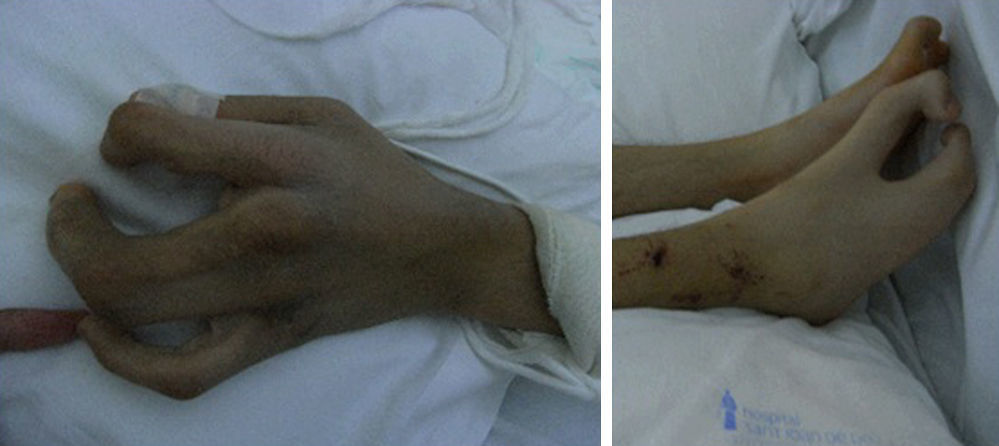
En ambas manos y pies presentaba ectrodactilia (fig. 1), con uñas estriadas. Mostraba alguna cicatriz de anteriores intervenciones de cirugía ortopédica, aunque con mínimas limitaciones funcionales. Se realizó una serie ósea que no mostró otras alteraciones excepto las derivadas del traumatismo.
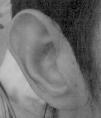
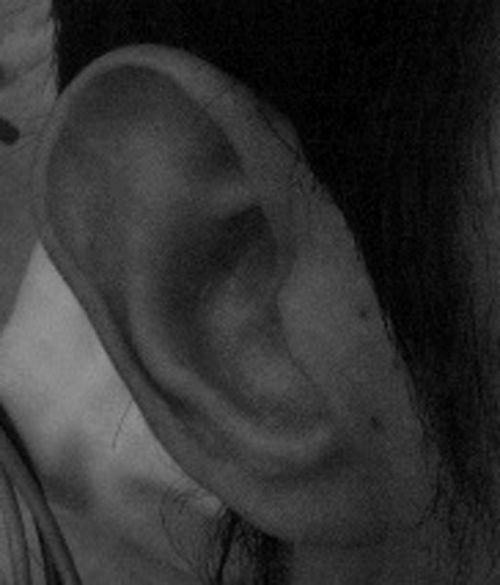
Había sido diagnosticado de obstrucción lacrimal congénita y presentaba epifora habitual, sin haber requerido tratamiento. Los pabellones auriculares eran dismórficos con forma de copa y de implantación baja (fig. 2). Intervenido de mastoiditis en una ocasión. Se realizaron potenciales auditivos de tronco cerebral, con resultado normal.
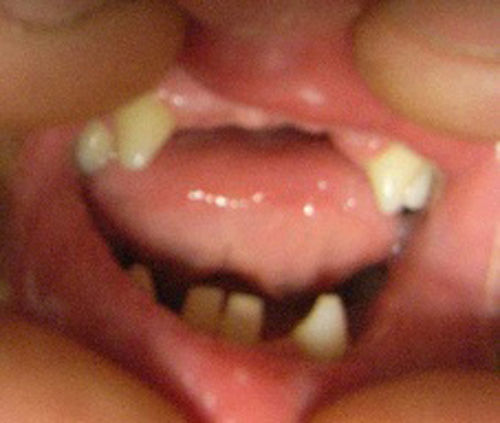
Presentaba alteraciones dentales (fig. 3), cambios de coloración, caries frecuentes, ausencia de algunas piezas dentarias. No presentaba fisura palatina.
Su rendimiento cognitivo global era dentro de la normalidad, sin relatar convulsiones en la infancia. Se descartaron también anomalías cardíacas y renales.
Se realizó estudio en los familiares, entre los que no había consanguinidad. En algunos miembros de la rama familiar materna (fig. 4) había distintos familiares afectados de ectrodactilia, con diversos grados de expresión clínica; además de varios afectados de fisura palatina y labial, agenesia de piezas dentales y alteraciones obstructivas en la vía lacrimal.
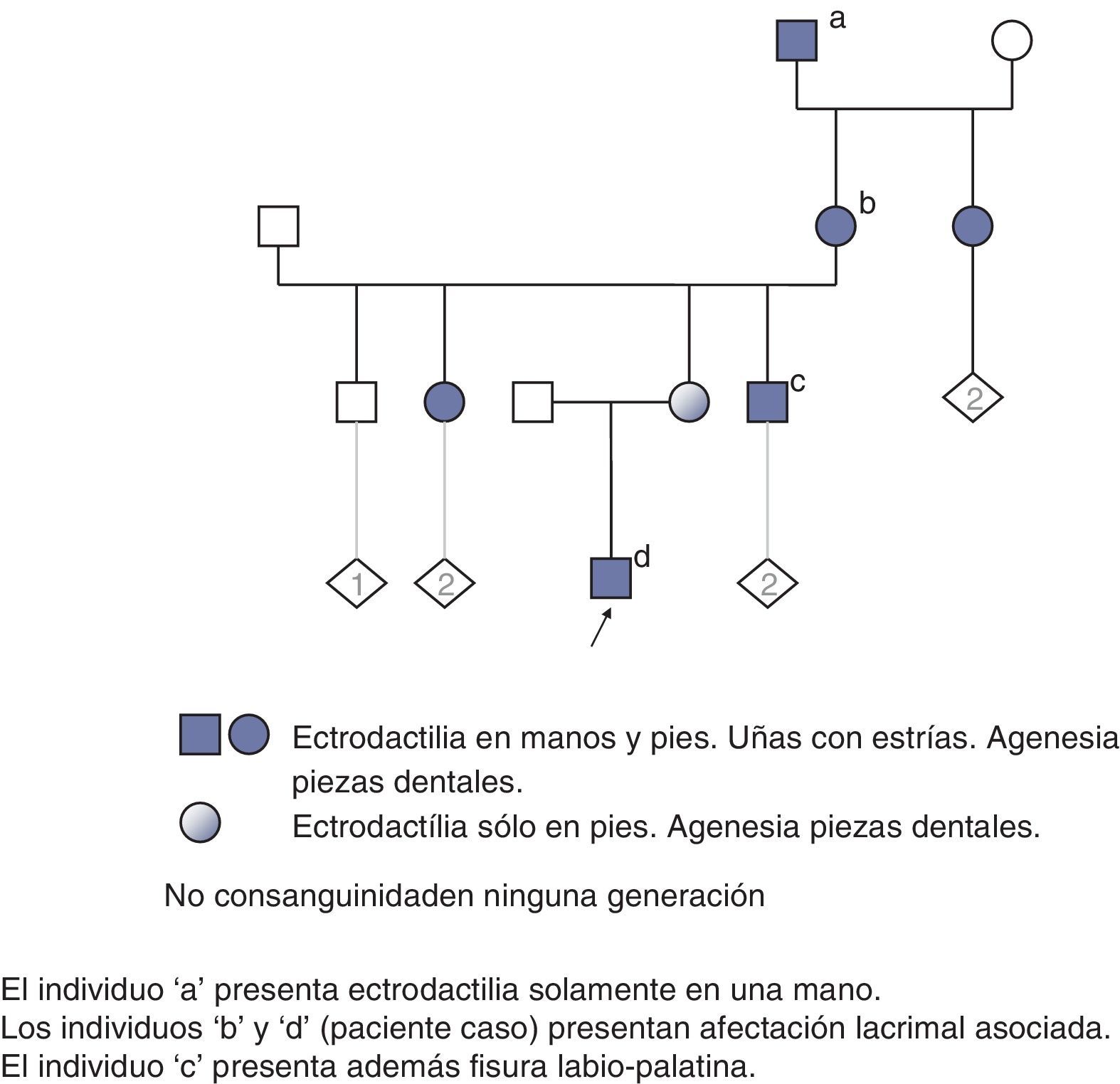
El estudio dismorfológico del paciente conjuntamente con la presencia de familiares afectos indicó el síndrome de Levy-Hollister como la entidad clínica más compatible.
DiscusiónEl paciente descrito representa un caso de síndrome de Levy-Hollister familiar, con expresividad variable y herencia autosómica dominante dentro de la misma familia. El diagnóstico es clínico y se basa en las alteraciones clínicas compatibles, conjuntamente con la agregación familiar correspondiente.
La frecuencia de este síndrome es desconocida y probablemente infravalorada, pues en muchos casos puede presentar una expresión casi silente. En 1967, Levy comunicó un caso de aplasia del conducto nasolacrimal en un paciente con dientes displásicos y alteraciones en las extremidades inferiores. En 1973, Hollister et al. describieron independientemente una familia con rasgos similares y propusieron una herencia autosómica dominante2,3. Desde entonces, se han descrito un centenar de casos, tanto esporádicos6,7 como familiares, todos ellos dando lugar a una herencia autosómica dominante con una expresividad variable.
El síndrome LADD se caracteriza por malformaciones lacrimales, pabellones auriculares de implantación baja y en forma de copa, diversos grados de sordera, agenesia/displasia dental y malformaciones en regiones distales de las extremidades1–8.
Las malformaciones lacrimales representan agenesia de los conductos lacrimales que normalmente se manifiestan con epifora1,6,8. La ausencia de esta puede orientar a una ausencia de glándula lacrimal unilateral, tal y como se ha descrito en la literatura8. La sordera puede ser de transmisión o neurosensorial, debido a malformaciones en el oído medio o interno, y se han descrito casos en los que esta es progresiva, por lo que muchos autores recomiendan exámenes audiométricos sucesivos6,9,10.
Además de las alteraciones dentarias, se ha descrito la ausencia de glándulas salivares, que predispone a más lesiones e infecciones dentales, así como la propia displasia o agenesia de las piezas dentales10.
Las malformaciones de partes distales de las extremidades suelen corresponder a los miembros superiores con una expresividad variable, pues se han descrito desde la ausencia de radio o la ectrodactilia hasta la clinodactilia del quinto dedo1,6,8.
En algunos casos se ha relacionado hipocalcemia, anomalías en las válvulas cardiacas y anomalías renales; en nuestro caso, no se ha evidenciado ninguna de éstas1–6,8. Se han descrito algunos casos de convulsiones e incremento del intervalo QT en el electrocardiograma11; no obstante, posteriormente se ha evidenciado la ausencia de glándulas paratiroideas, con hipoparatiroidismo primario e hipocalcemia, que una vez corregida ha supuesto la normalización de la clínica12,13.
La etiología se ha relacionado con mutaciones en los dominios tirosincinasa en los genes que codifican los receptores de los factores de crecimiento de fibroblastos 2 y 3 (FGFR2 y FGFR3). También se han descrito mutaciones en el factor de crecimiento 10 de los fibroblastos (FGF10). Existe gran heterogeneidad genética y clínica en los casos descritos8,14–16.
Queremos destacar la importancia del diagnóstico clínico-dismorfológico en un paciente ingresado en la UCIP por causas no relacionadas con la afección de base. El estudio posterior permitió llegar a un diagnóstico de síndrome de Levy-Hollister familiar y posteriormente a un asesoramiento dismorfológico familiar por el departamento de genética clínica.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
