





Son muchos los genes que se han implicado en la diferenciación testicular, cuyas alteraciones dan cuadros de trastornos de la diferenciación sexual y cariotipo 46XY.
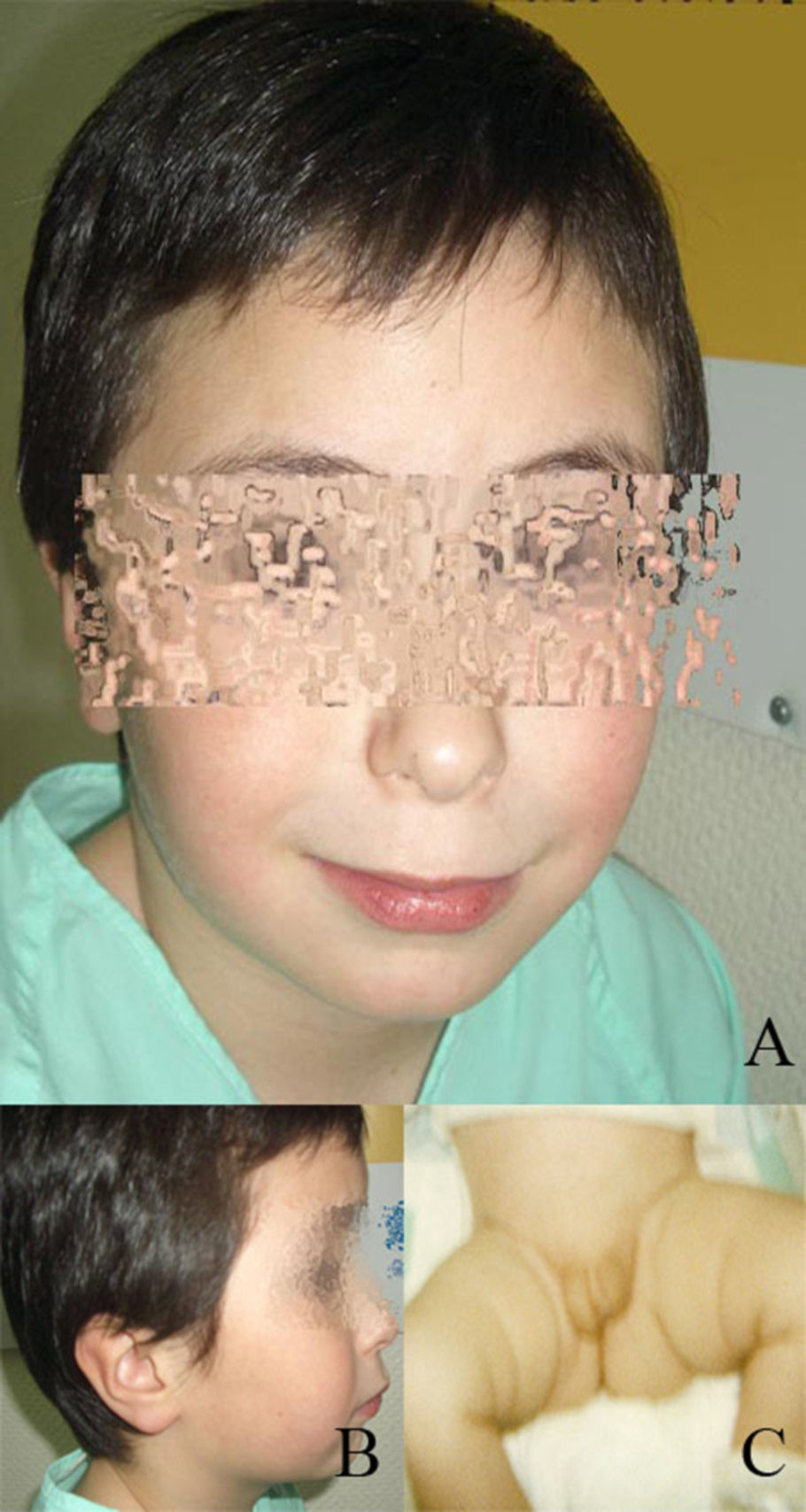
Caso clínicoRecién nacido con hipospadias interescrotal, gónadas palpables y pene hipoplásico. Cariotipo 46XY. Ecografía abdominal: testes y sin restos müllerianos. Buena respuesta al test corto de gonadotropinas. Al año presenta retraso psicomotor, hipotonía. Resonancia magnética con atrofia de sustancia blanca frontotemporal y disminución del cuerpo calloso. Biopsia testicular compatible con disgenesia gonadal. Dada la situación intersexual al nacimiento, el retraso psicomotor y la presencia de dismorfias faciales se solicita cariotipo de alta resolución: deleción 46, XY, del(9p)(p23-pter).ish tel (9p-).
ComentariosSon muchos los genes implicados en la diferenciación testicular, algunos de ellos también influyen sobre el desarrollo de otros tejidos. En el brazo corto del cromosoma 9 se encuentran dos genes, DMRT1 y DMRT2, implicados en la diferenciación sexual, cuyas alteraciones también han sido descritas como causantes de retraso mental. En la evaluación de los trastornos de la diferenciación sexual son muy importantes los signos acompañantes para poder orientar el estudio genético.
Many genes are involved in testicular differentiation. The alterations of these genes are responsible for sexual differentiation disorders with 46 XY karyotype.
CaseWe report the case of a newborn who had an interscrotal hypospadias, palpable gonads and hypoplastic penis. Karyotype 46 XY. Abdominal ultrasound revealed testes and absence of Müllerian remnants. There was a good response to the short gonadotrophin test. At one year he had signs of psychomotor retardation and hypotonia. The magnetic resonance revealed frontal-temporal atrophy and a decrease in the corpus callosum. Testicular biopsy was compatible with gonadal dysgenesis. A preoperative cystography showed a vaginal remnant. Due to the presence of a sexual differentiation disorder, psychomotor retardation and facial dysmorphism, we requested a high-resolution karyotype: deletion 46, XY, del (9p) (p23-pter). Ish tel (9p-).
DiscussionMany genes are involved in testicular differentiation, some of which also affect the development of other tissues. In the short arm of chromosome 9, two genes, DMRT1 and DMRT2, are involved in sexual differentiation. Their alterations have also been described as causing mental retardation. In the evaluation of 46,XY disorders of sex differentiation, the accompanying signs are very important for guiding the genetic study.
El desarrollo de las gónadas masculinas y femeninas, así como de los genitales externos, es el resultado de interacciones genéticas y hormonales. El gen SRY, localizado en el cromosoma Y, actúa directamente sobre la gónada bipotencial activando la diferenciación testicular1. Estudios posteriores han demostrado la importancia de otros genes involucrados en la diferenciación sexual, como el gen WT1 en el cromosoma 11p132, el gen SF-1 en el 9q333, el gen SOX-9 en el 17q24.3-q25.14, el gen DAX-1 en el cromosoma Xq225 y otros muchos también publicados. No en todos los casos presentan fenotipo completamente femenino, sino que muchas veces se trata de ambigüedad genital más o menos marcada. Aún así, son muchos los casos de trastornos de la diferenciación sexual por disgenesia gonadal, con cariotipo 46XY, que permanecen sin explicación genética conocida, lo que sugiere la existencia de más genes que están aún por descubrir.
Presentamos un caso de trastorno de la diferenciación sexual, disgenesia gonadal, y retraso mental leve en un paciente con deleción 46,XY,del(9p)(p23-pter).ish tel(9p-). Existen trabajos previos que muestran pacientes con fenotipo femenino o ambigüedad genital, con cariotipo 46 XY y alteraciones en el brazo corto del cromosoma 96,7. Los trastornos en el comportamiento, retraso mental y otras alteraciones también han sido descritos en pacientes afectos por alteraciones de esta región8–10.
Caso clínicoRecién nacido que presenta hipospadias interescrotal con palpación de ambas gónadas en labios escrotales, pene hipoplásico, de 2cm, con meato estrecho (estadio III de Prader) (fig. 1). El embarazo fue controlado, sin ingesta de medicación. Cariotipo en líquido amniótico: 46 XY. Parto a término, eutócico. PRN: 3.390g (p=50), LRN: 52cm (p=80), P. Cef.: 36’8cm (p=85). Cribado metabólico normal. En la historia no se obtienen antecedentes familiares de interés, salvo un tío paterno con extrofia vesical.
Se solicita un nuevo cariotipo 46 XY, 17 hidroxiprogesterona 2,9ng/ml, androstendiona 4,4ng/ml y renina 5,7ng/ml. Se practica ecografía abdominal en la que no se visualiza imagen de útero ni de vagina y sí se observan imágenes de testes en escroto. La existencia de tejido testicular se confirma con la respuesta al test corto de gonadotropinas (2.500UI HCG intramuscular en dosis única): testosterona a las 72h de 2,317pg/ml. Con todos estos datos se prosigue el estudio con el gen de receptores de andrógenos y la 5 alfa reductasa, que resultan normales.
Con el fin de aumentar el tamaño del pene y lograr mayor virilización, se inicia a los 4 meses tratamiento con cipionato de testosterona observándose el aumento del pene hasta 3,5cm.
En su evolución se observa retraso psicomotor a partir del año, con marcada hipotonía generalizada. Se realiza resonancia magnética en la que se objetiva leve hipoplasia de vermis inferior, con amplia comunicación del cuarto ventrículo con la cisterna magna (probable variante de Dandy Walker), supratentorialmente hay signos de de atrofia de predominio frontotemporal bilateral, a expensas de sustancia blanca, con leve disminución del tamaño del cuerpo calloso.
A los dos años, se interviene de hipospadias severo y criptorquidia bilateral. Se obtiene biopsia de ambos testículos que revela la existencia de tejido testicular. A nivel de la albugínea, presenta pérdida de la colagenización con presencia de túbulos seminíferos en su espesor. El diámetro tubular está ligeramente disminuido, el número de células de Sertoli y de células germinales es bajo. No se observan células tumorales. Esta biopsia es compatible con disgenesia gonadal.
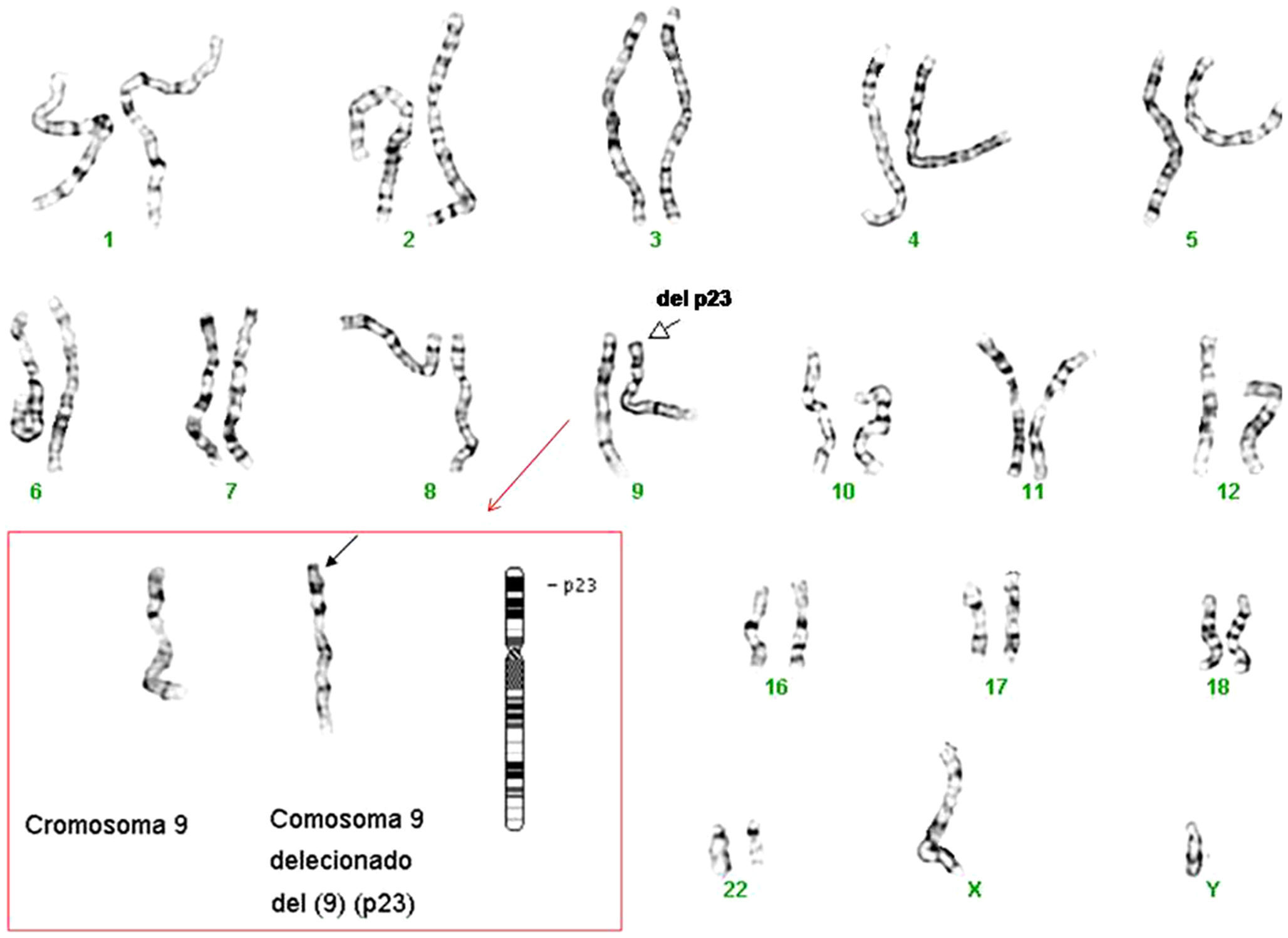
Dada la situación intersexual al nacimiento, el retraso psicomotor y la presencia de dismorfias faciales que se van haciendo cada vez más marcadas, como hipertelorismo, hipoplasia malar, epicantus, labio superior fino, etc. (fig. 1), y se solicita cariotipo de alta resolución. El estudio mostró 46 cromosomas con fórmula sexual XY. Con las técnicas de bandas G de alta resolución (550–850 bandas), se observó que uno de los cromosomas del par 9 era anómalo. Con la aplicación de la técnica de citogenética molecular (Cromoprobe Multiprobe-T System, Cytocell) se observó una deleción del telómero de brazo corto del cromosoma 9. Finalmente, con las técnicas de bandas G se determinó la deleción terminal de las bandas (p23-pter) de un cromosoma del par 9. El cariotipo es por tanto: 46,XY,del(9p)(p23-pter).ish tel(9p-) (fig. 2). Dado que el estudio de cariotipo de alta resolución de los padres es normal, la alteración de nuestro paciente es «de novo».
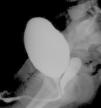
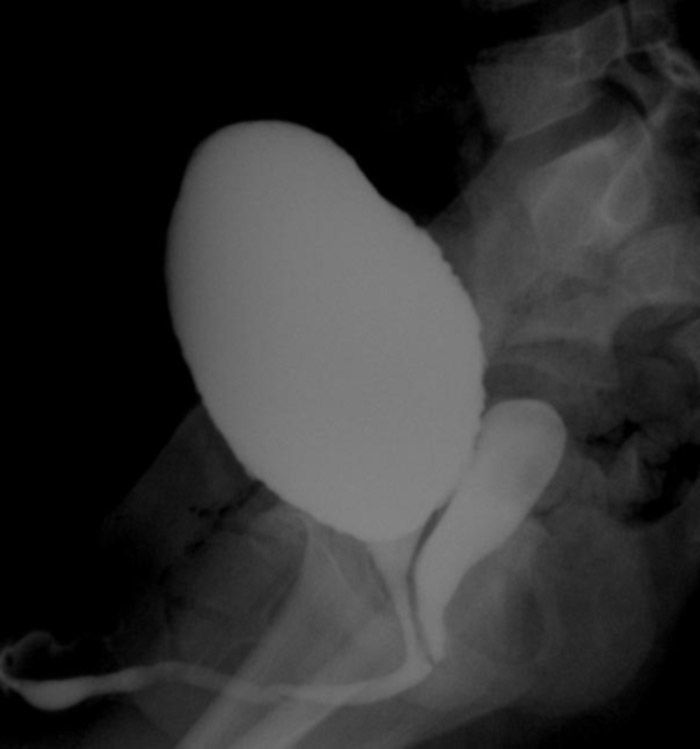
A los 4 años de edad, presenta fistulización del hipospadias que requiere nueva intervención. En el estudio previo se hace cistografía que revela la existencia de vejiga y uretra de características masculinas, con una cavidad compatible con vagina que desemboca en uretra (fig. 3). En la extirpación de dicha cavidad se obtiene biopsia compatible con restos mullerianos.
En la última revisión, con 7 años, la madre refiere que tiene cada vez más problemas para centrarse en las tareas, con dificultad en el aprendizaje y necesidad de recibir apoyo escolar.
DiscusiónLos pacientes con cariotipo 46XY y trastornos de la diferenciación sexual tienen una incidencia desconocida. La presencia de genitales externos ambiguos y restos de conductos de Müller nos indica el fallo en el desarrollo testicular con alteración en la producción de andrógenos y de factor inhibidor mulleriano (MIF). Son muchos los genes que se han implicado en la diferenciación testicular, lográndose su identificación en el 70% de las ocasiones. Algunos de ellos también influyen sobre el desarrollo de otros tejidos (tabla 1).
Alteraciones genéticas asociadas a trastornos de la diferenciación testicular
| Gen | Localización | Año/autor | Manifestaciones |
| SRY | Yp11 | 1990/Sinclair | Disgenesia gonadal |
| DAX1 | Xp21 | 1994/Bardoni | Disgenesia gonadal (si duplicación) |
| Hipoplasia adrenal congénita e hipogonadismo hipogonadotropo (si mutación inactivadora) | |||
| DMRT1 | 9p24.3 | 1998/Raymond | Disgenesia gonadal |
| Retraso mental | |||
| DMRT2 | 9p24.3 | 2000/Ottolenghi | Disgenesia gonadal |
| Retraso mental | |||
| SF1 | 9q33 | 1994/Luo | Disgenesia gonadal |
| Hipoplasia adrenal congenital | |||
| Hipogonadismo hipogonadotropo | |||
| WT1 | 11p13 | 1990/Call | Disgenesia gonadal |
| Tumor de Wilms | |||
| SOX9 | 17q24 | 1994/Foster | Disgenesia gonadal |
| Displasia campomiélica | |||
| – | 10q | 1993/Wilkie | Disgenesia gonadal |
El síndrome de monosomía del 9p, síndrome de Alfi, está caracterizado por retraso mental y anomalías somáticas (trigonocefalia, hipertelorismo, hipoplasia malar, epicantus, fisuras palpebrales pequeñas, carinas antevertidas, microstomía y micrognatia) y tiene una incidencia aproximada de 1/50.000 recién nacidos11.
Aunque los trastornos en la diferenciación sexual por alteraciones del cromosoma 9 están descritos por Huret et al desde 1988, no es hasta 1999 cuando se describen las deleciones en el brazo corto del cromosoma 9 y se sugiere la presencia de los genes DMRT1 y DMRT2. Estos genes fueron propuestos como responsables de dicha anomalía, debido a su gran similitud con las secuencias Doublesex y mab-3, reguladores de la diferenciación sexual en la Drosophila y en Caenorhabditis12.
Existen varias teorías acerca del mecanismo por el que la monosomía del 9p24 causa disgenesia gonadal. La explicación más aceptada es que la haploinsuficiencia del 9p24 detiene la diferenciación testicular. Esta es la misma teoría que Wagner et al propusieron para la disgenesia gonadal causada por alteraciones del SOX94, así como Washburn y Eicher hicieron lo propio con el cromosoma 17 y sus estudios en ratones13.
El retraso mental, la dificultad para el aprendizaje y las alteraciones del comportamiento son características descritas en niños con mutaciones del 9p-8,10,14. Un niño con craneosinostosis, retraso psicomotor y alteración del cuerpo calloso fue descrito por Eshel et al15. Es importante destacar que alteraciones en el cromosoma 9p también han sido descritas como causantes de retraso mental y alteraciones del comportamiento en mujeres con cariotipo 46XX y varones con cariotipo 46XY con desarrollo sexual normal. Esta última observación, es la que ha hecho plantearse si la disgenesia gonadal con espectro autista es una única entidad o se trata de entidades genéticamente diferentes. En esta última dirección se encuentra la demostración de los múltiples genes (DOCK8, COBW, ANKRD15 y FOXD4) localizados en el brazo corto del cromosoma 9, que pueden ser responsables de los trastornos del comportamiento, lenguaje y aprendizaje16,17.
Otro punto importante que hay que destacar es la existencia de gonadoblastoma en pacientes con disgensia gonadal por alteraciones en el 9p. En general, los gonadoblastomas son exclusivos de disgenesias gonadales en las que existe cromosoma Y o al menos un fragmento, siendo la incidencia aproximadamente de un 30%. Hasta la fecha son 4 los pacientes con deleciones en el brazo corto del cromosoma 9 descritos en la literatura con gonadoblastoma, lo que representa un 15–20%7,9,18,19. En nuestro caso, la biopsia testicular no reveló signos tumorales, planteándose la discusión sobre si debe realizarse gonadectomía, como aconsejan Gibbons y Müller en sus respectivos trabajos19,20, o, por el contrario, se deben seguir controles periódicos vigilando signos de malignización.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
