

El complejo esclerosis tuberosa (CET) es una enfermedad genética autosómica dominante que afecta a 1/6.000 nacidos vivos1–3. Suele ocurrir de novo en dos tercios de los casos y se caracteriza por el desarrollo de hamartomas en cerebro, piel, riñones, ojos, corazón, pulmones e hígado1,2. Se asocia a menudo con trastornos de conducta, convulsiones y déficit cognitivo3,4. En el 75-85% de los pacientes con CET se halla una mutación en los genes TSC1 o TSC2, siendo éste último el más frecuente1,4.
Presentamos un análisis retrospectivo del fenotipo clínico y genotipo de los pacientes con diagnóstico clínico de CET desde 1983 hasta 2009 en nuestro centro.
Se estudiaron 16 pacientes (tabla 1), de los que 10 (62,5%) eran de sexo femenino. La edad media de diagnóstico fue de 4,4 meses. El síntoma inicial más frecuente (68,7%) fue la epilepsia. La mayoría de los pacientes (60%) presentaron un nivel cognitivo normal y el 53,3% fueron diagnosticados de trastorno de conducta (3 conducta autista, 4 hiperactividad).
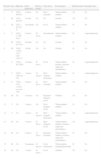
Características fenotípicas y mutaciones identificadas en cada uno de los pacientes
| Paciente | Sexo | Mutación | Edad diagnóstico | Retraso mental | Crisis inicio | Neuroimagen | Rabdomiomas | Patología renal |
| 1 | V | TSC1. R245X | 6 meses | No | West (resuelto) | Tuberosidades; nódulos | No | No |
| 2 | M | TSC1. R245X | 33 años | No | No | Normal | No | No |
| 3 | M | TSC 2. R1138 X | Nacimiento | No | Focal | Tuberosidades; nódulos | Sí | No |
| 4 | V | TSC2. C1286 del A | 8 meses | Sí (leve) | Generalizada | Tuberosidades; nódulos | Sí | Angiomiolipoma |
| 5 | M | TSC2. E1337X | Prenatal | No | No | Normal | Sí | No |
| 6 | M | TSC2. c4346-4347 insG exón 33 | 16 días | No | No | Normal | Sí | No |
| 7 | V | TSC2. R1720W | 18 meses | Sí (leve) | Focal | Tuberosidades; nódulos; trastorno migración neuroblástica | Sí | Angiomiolipoma |
| 8 | V | TSC2. 2092 ins CG | 5 meses | Sí (grave) | West (después refractaria) | Tuberosidades; nódulos | Sí | Angiomiolipoma |
| 9 | V | TSC2. 3832 +1G>C en exón 30A | 8 meses | Sí (leve) | West (resuelto) | Tuberosidades; nódulos | No | No |
| 10 | M | No | 10 meses | No | Espasmos (después focal) | Nódulos | No | No |
| 11 | V | No | 5 meses | No | West (después focal) | Tuberosidades; nódulos | Sí | No |
| 12 | V | No | 3 meses | Sí (grave) | West (después refractaria) | Tuberosidades; nódulos | No | Angiomiolipoma |
| 13 | M | No | 4 meses | Sí (leve) | West (después focal) | Tuberosidades; nódulos; trastorno migración neuroblástica | No | Angiomiolipoma |
| 14 | M | No | 9 meses | Sí (leve) | Espasmos (resuelto) | Tuberosidades; nódulos; trastorno migración neuroblástica | Sí | No |
| 15 | M | No | Nacimiento | Sí (grave) | Focal (refractaria) | Tuberosidades hemimegalencefalia | Sí | No |
| 16 | M | No | 7 meses | Sí (leve) | Focal | Nódulos | No | No |
Signos cutáneos: máculas hipomelanóticas (15); angiofibromas o placa frontal (9). Alteraciones en la neuroimagen: nódulos subependimarios (12), tuberosidades corticales (11) y alteraciones de migración neuroblástica (4). La figura 1 es una imagen de RM de uno de los pacientes.
 en la que se aprecian túberes corticales y nódulos subependimarios.")
Los rabdomiomas cardíacos se diagnosticaron en 9 pacientes (56,3%), siendo la mayoría asintomáticos. Los angiomiolipomas renales aparecieron en 5 (35,7%).
Al igual que han reflejado diversos autores2–5, la mayoría de los pacientes (81,2%) presentaron epilepsia. Las crisis tienen un origen focal o multifocal, con correspondencia entre el foco electroencefalográfico y las lesiones hiperintensas en la RM, lo que demuestra la importancia de los túberes corticales como focos epileptógénos5. Se ha escrito mucho sobre la importancia del sistema inhibidor GABA-érgico en las crisis epilépticas de los pacientes con CET, confirmado por la particular eficacia terapéutica de la vigabatrina, inhibidor de la transaminasa GABA (50% de los niños con CET y espasmos infantiles responden a este fármaco6).
En nuestros pacientes, la epilepsia fue fácil de controlar en la mayoría de los casos, aunque casi la mitad presentaron síndrome de West en algún momento de su evolución, algo más que en otras series, en las que aparece en torno al 25%4,5. Los fármacos más utilizados fueron vigabatrina y valproato.
En cuanto a la afectación cognitiva, existe una amplia variabilidad interindividual, alrededor del 45% presenta algún grado de deterioro y un 30% del total tiene un retraso mental grave7. De los pacientes con inteligencia normal, la mayoría presentarán algún grado de dificultad de aprendizaje, trastorno del lenguaje, de conducta o déficit atencional5,7. Un 40% de nuestros pacientes presentaron dificultades de aprendizaje y los casos de trastorno de conducta fueron frecuentes (53%), siendo diagnosticados 3 de ellos de trastorno del espectro autista.
Algunos trabajos recientes han relacionado la presencia de mutaciones en el gen TSC2 con un fenotipo más grave, mayor probabilidad de trastorno de conducta, epilepsia y retraso mental1,4,5. En nuestra pequeña serie, 4 de los 7 pacientes con mutación TSC2 identificada (57,1% del total de los TSC2) presentaban retraso mental y epilepsia. Aunque nuestra muestra no es representativa por diversos motivos (es pequeña, el estudio genético no se ha realizado a todos los pacientes, el seguimiento no ha sido sistemático en todos los casos), esta cifra es algo inferior de lo que cabría esperar. Estudios más amplios y sistemáticos podrían en el futuro comprobar esta afirmación en la población infantil española con CET.
