


Sr. Editor:
Las malformaciones oculares congénitas, aunque menos frecuentes que otros defectos congénitos, suelen ser anomalías incapacitantes. Su diagnóstico clínico al nacimiento puede resultar dificultoso en muchas ocasiones, debido a diferentes causas, como el edema palpebral, que dificultan la correcta exploración ocular al nacimiento, por lo que es difícil obtener datos epidemiológicos sobre los mismos. Se estima una incidencia global del 3,68/10.000 nacidos vivos, siendo los más frecuentes la catarata congénita (6,31/10.000) y los colobomas en todas sus variedades, tanto de globo ocular como de sus anejos (4,89/10.000) 1,2. El coloboma palpebral es un defecto congénito extremadamente raro, del que sólo se ha encontrado un caso publicado en la revisión bibliográfica realizada 1,3,4.
Presentamos el caso de una niña recién nacida a término, de una gestación de 40 semanas, progenitores jóvenes, no consanguíneos. Embarazo sin antecedentes de metrorragias o hipertensión arterial, ni de exposición a posibles teratógenos conocidos y cuyos controles ecográficos fueron normales. La amniocentesis aportó un cariotipo normal. Parto vaginal, eutócico, test de Apgar de 10 al min y a los 5 min y un peso de 2.950 kg. Tras el nacimiento se observa una hendidura en ambos párpados superiores, de localización centro-nasal (fig. 1), con los bordes bien definidos, de aproximadamente 1 cm de amplitud, que deja al descubierto la correspondiente zona de córnea (fig. 2). Se diagnostica de coloboma palpebral bilateral, no encontrando otros hallazgos en la exploración física.
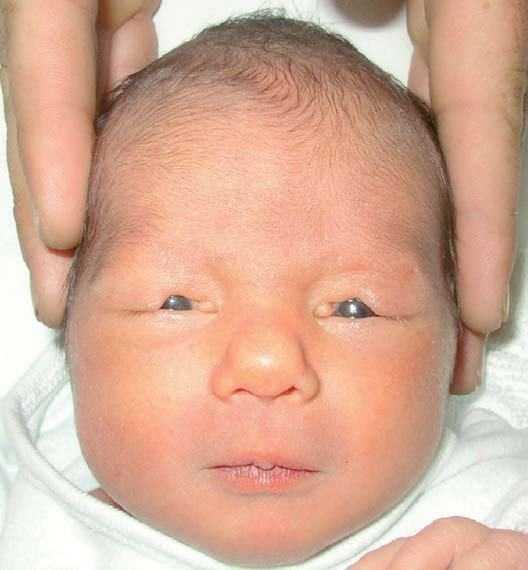
Figura 1. Coloboma palpebral de localización centro-nasal en ambos párpados superiores.
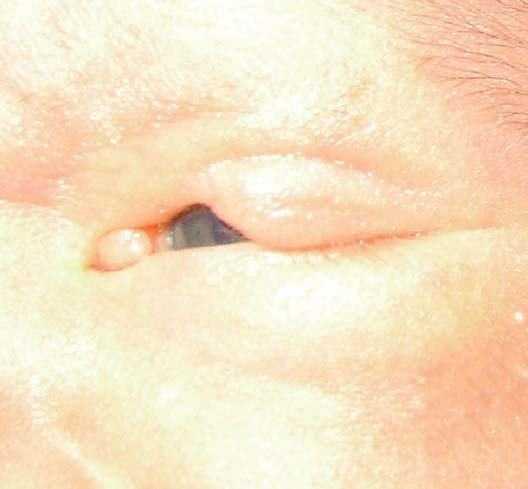
Figura 2. Detalle del coloboma con el párpado cerrado, en el que se aprecia la zona de córnea expuesta y los bordes bien definidos.
Como exploraciones complementarias se solicitan una ecografía ocular, cerebral y abdominal/renal, para descartar otras malformaciones derivadas de tejido ectodérmico asociadas, así como valoración por el Servicio de Oftalmología Pediátrica, que le realiza examen ocular y fondo de ojo, resultando normales.
Actualmente tiene 20 meses de vida, ha completado normalmente los hitos del desarrollo, y la exploración física sigue siendo totalmente normal con excepción del coloboma palpebral.
La niña ha permanecido asintomática en todo momento y con medidas de lubricación ocular (lágrimas artificiales, pomada epitelizante y cámara de humedad), manteniendo una correcta hidratación ocular hasta el momento de la intervención, que se realiza al año de vida, con excelentes resultados estéticos y funcionales, no requiriendo ningún tipo de tratamiento tras la intervención quirúrgica, siendo dada de alta al año y medio de vida.
Para el correcto crecimiento y formación de los párpados y sus estructuras marginales, se requiere una completa adhesión epitelial entre las distintas capas palpebrales en la 9.ª semana y su mantenimiento entre la 10.ª semana y el 6.º mes 1,3. El origen del coloboma, estaría probablemente en anomalías en el sistema vascular o en el desarrollo de las células de la cresta neural, implicadas en los síndromes del primer arco branquial en general y en la fusión, en algunos casos asociado con bandas fibrosas anormales que causarían fisuras faciales 3,4.
Los colobomas afectan principalmente al margen palpebral. Son más frecuentes en el párpado superior, generalmente unilaterales (raro bilaterales) y en localización nasal. Cuando están en el párpado inferior, con frecuencia este se encuentra ausente en todo su grosor, aunque también puede presentar los bordes redondeados y cubiertos de conjuntiva o bien, en casos mucho menos frecuentes, puede existir ausencia exclusiva del tarso 4,5.
Al hablar de sus posibles asociaciones (recordemos que el grupo de los colobomas en general era el que más frecuentemente se asociaba a múltiples anomalías congénitas), hemos de nombrar tanto las oculares, como las sistémicas. Entre las oculares destacan el dermoide, el lipodermoide, el queratocono, el coloboma de iris y la microftalmía 4,5. En cuanto a los sistémicos, en su mayoría síndromes de primer y segundo arcos branquiales, es de destacar que los de localización en el tercio externo del párpado inferior se asocian con mayor frecuencia al síndrome de Treacher-Collins (MIM: 154500), que asocia estos colobomas hasta en un 69 % de las ocasiones, cuya expresión es muy variable, aunque se presume como autonómica dominante, en el 60 % de los casos ocurre debido a mutaciones espontáneas (se ha identificado una concreta situada en 5q32-33.1, como responsable de algunos casos) y la disostosis craneofacial 6.
Los de localización en el párpado superior, con relativa frecuencia se asocian con el síndrome de Goldenhar/Espectro facio-aurículo-vertebral (MIM: 164210), que a diferencia del anterior, presenta mayor incidencia y etiología desconocida, pero en el que el coloboma palpebral no se incluye con tanta frecuencia entre su amplio espectro de manifestaciones. También debemos nombrar el síndrome del coloboma-lipoma nasopalpebral (MIM: 167730), de localización, como indica su nombre a nivel nasal y que se transmite con herencia autosómica dominante, con penetrancia completa, así como los diversos síndromes de disostosis acrofacial (MIM: 263750) 4,5,7.
En su evolución es relativamente frecuente la elevada incidencia de estrabismo, defectos de refracción, opacidades y bandas fibróticas, que limitan la movilidad ocular, lo que refuerza la importancia de la protección ocular de modo profiláctico desde el momento de su diagnóstico 8.
Al hablar del tratamiento, de un modo general, la mayoría de los autores apoyan la reparación precoz en caso de exposición corneal total para prevenir cicatrices corneales y en caso de no existir tal exposición, posponer la reparación hasta los 2-3 años 8,9. Actualmente con las nuevas técnicas quirúrgicas, se elige el tratamiento en función de la extensión del defecto, de modo que en colobomas pequeños, se prefiere el tratamiento conservador con lubricantes, cámaras de humedad herméticas o parches temporales nocturnos hasta la realización de cirugía correctiva cuando los tejidos maduren. En cuanto a la cirugía, en defectos menores del 35 %, se realiza cirugía de cierre directo, mientras que en defectos mayores, entre el 35-45 % de extensión, se realiza cantotomía lateral y en los de extensión mayor al 45 % se prefiere la reconstrucción en 2 tiempos 8-10.
Correspondencia: Dr. A. Sánchez Andrés.
Servicio de Pediatría. Hospital Universitario La Fe.
Avda. de Campanar, 11. 46009 Valencia. España.
Correo electrónico: llanosytoni@ono.com
