

Los cariotipos 48,XXXY y 49,XXXXY representan aneuploidías infrecuentes de los cromosomas sexuales, considerados clásicamente como variantes del síndrome de Klinefelter (SK) (47,XXY), dado que comparten la disgenesia testicular y el hipogonadismo hipergonadotropo. Sin embargo, la mayor frecuencia de alteraciones endocrinológicas, ortopédicas y neuropsicológicas los diferencia de la forma 47,XXY1.
Presentamos un varón de 9 años y 6 meses con mosaico 48,XXXY/49,XXXXY diagnosticado a los 4 meses de vida, en el contexto de hipotonía generalizada y retraso psicomotor.
Padres sanos, no consanguíneos. Antecedentes personales: parto a término (38+2 semanas) con peso al nacimiento de 2,860g (−0,71 DE), longitud de 47cm (−1,12 DE) y perímetro cefálico de 35cm (+0,09 DE). Apgar 7/9. Genitales normales. Cribado metabólico y auditivo normal. Seguimiento cardiológico por foramen oval permeable y comunicaciones interventriculares apicales múltiples, con resolución espontánea. Caries y flemones dentales recurrentes. Retraso psicomotor grave (cociente intelectual<50): sonrisa social al mes de vida, sostén cefálico a los 6 meses, sedestación a los 2 años, deambulación a los 3 años. Lenguaje escaso, repetitivo, con dislalias, con mayor limitación a nivel expresivo que comprensivo. Velocidad de crecimiento normal, siguiendo el percentil 50-75 de talla y 90-97 de peso, con edad ósea retrasada aproximadamente un año.
Exploración física: peso 37,8 kg (+2,77 DE), talla 136,6cm (+0,23 DE), IMC 20,3 kg/m2 (+1,44 DE), perímetro cefálico 50cm (−2,21 DE) y braza 129,5cm. Talla diana 167,3±5cm (−1,36 DE). Ptosis palpebral, epicantus, fisuras palpebrales mongoloides, raíz nasal ancha, orejas prominentes, prognatismo, caries múltiples, microcefalia y cabello de implantación baja. Limitación a la pronosupinación de ambos antebrazos, con imposibilidad de superar la posición neutra. Quinto metacarpiano corto. Genu valgus, pies planos con sobreposición del quinto dedo. Auscultación cardiopulmonar normal. Tanner I (G1P1Aa), testes de 1ml, pene de 3,4×1,4cm (−2,9 DE), sin ginecomastia.
Exploraciones complementarias: hormonas tiroideas, FSH, LH, testosterona, estradiol, inhibina B y hormona antimulleriana normales. Metabolismo de hidratos de carbono y lipidograma sin alteraciones. Radiografía de antebrazos: sinóstosis radioulnar proximal (fig. 1). Resonancia magnética craneal: lesiones de sustancia blanca de predominio frontal y dilataciones periventriculares de Virchow Robin, compatibles con leucoencefalopatía inespecífica. Cariotipo: 90% de las células 49,XXXXY y 10% 48,XXXY.
 Visión frontal y lateral del paciente. C) Radiografía de antebrazos con hallazgo de sinóstosis radioulnar.")
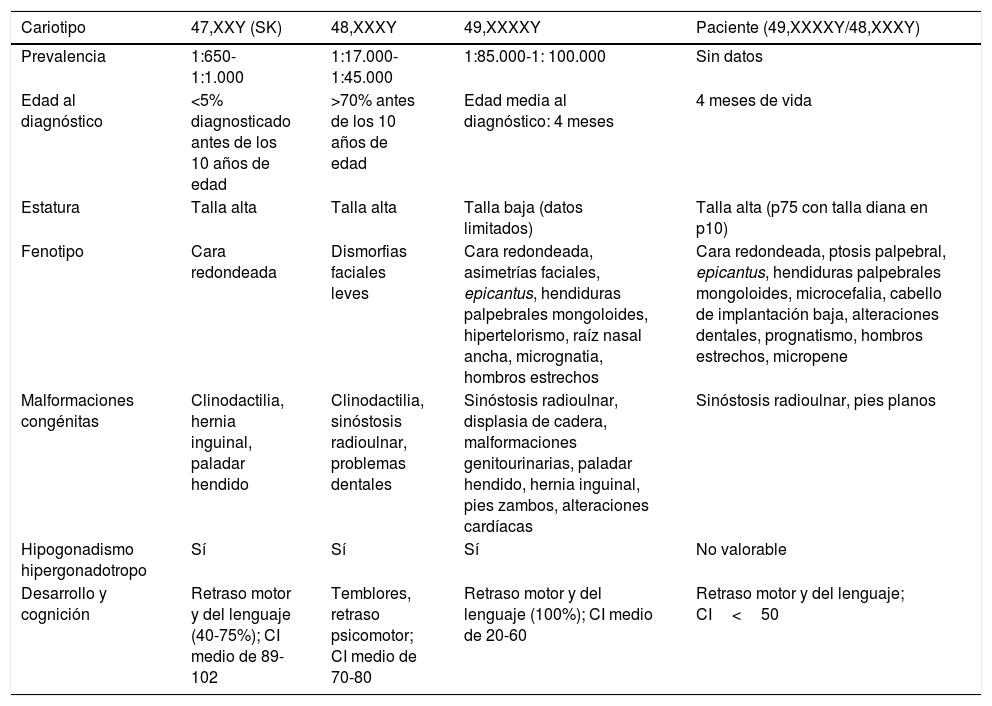
La incidencia de las variantes 48,XXXY y 49,XXXXY es mucho menor en comparación con el SK, 1:17.000-1:50.000 y 1:85.000-1:100.000 nacidos vivos, respectivamente1,2. Presentamos una forma muy infrecuente de mosaico 48,XXXY/49,XXXXY, de incidencia desconocida. Mientras que el SK se asocia a talla alta al igual que la variante 48,XXXY, la forma 49,XXXXY presenta talla baja, por razones que aún se ignoran. Nuestro caso presenta talla normal, aunque alta para su talla diana. La mayoría de las características previamente descritas son comunes a ambas variantes y a la forma 47,XXY2 (tabla 1). La frecuencia de sinóstosis radioulnar aumenta a mayor número de cromosomas X adicionales2. Por tanto, aunque se han descrito en mosaicos 47,XXY/46,XX, son más frecuentes en las variantes con tetrasomía y pentasomía3. Otras malformaciones congénitas como paladar hendido, displasia renal y de cadera o hernias inguinales no han sido halladas en nuestro paciente, pese a ser más frecuentes y severas en la forma 49,XXXXY1,4. El hipogonadismo puede presentarse como micropene, criptorquidia, hipoplasia escrotal o hipogonadismo hipergonadotropo. Este último es un hallazgo común al SK y sus variantes. Nuestro paciente en edad prepuberal presenta un volumen testicular de 1ml y micropene, otro hallazgo común a todas las formas1. En las formas 47,XXY y en las variantes con tetrasomía y pentasomía, existe un amplio espectro de disfunción cognitiva; la forma 49,XXXXY es la de mayor afectación1,4 (tabla 1). Nuestro paciente presenta retraso psicomotor grave con importante limitación del lenguaje expresivo, hecho común a todas las formas. Tartaglia et al. describen retraso motor e hipotonía en el 100% de los individuos 49,XXXXY, con inicio de la deambulación a una edad media de 25,5 meses1. Visootsak et al. estiman un descenso del cociente intelectual de 10-15 puntos por cada cromosoma X adicional2,5. Las anomalías del parénquima cerebral (ventriculomegalia, hiperintensidades de la sustancia blanca) y de la unión cráneo-cervical también se asocian a las variantes de SK, como describen Milani et al.6. A excepción de lesiones inespecíficas de la sustancia blanca, dichas anomalías no se han objetivado en nuestro paciente.
Características clínicas de las variantes cromosómicas 47,XXY; 48,XXXY; 49,XXXXY y del paciente 48,XXXY/49,XXXXY
| Cariotipo | 47,XXY (SK) | 48,XXXY | 49,XXXXY | Paciente (49,XXXXY/48,XXXY) |
|---|---|---|---|---|
| Prevalencia | 1:650-1:1.000 | 1:17.000-1:45.000 | 1:85.000-1: 100.000 | Sin datos |
| Edad al diagnóstico | <5% diagnosticado antes de los 10 años de edad | >70% antes de los 10 años de edad | Edad media al diagnóstico: 4 meses | 4 meses de vida |
| Estatura | Talla alta | Talla alta | Talla baja (datos limitados) | Talla alta (p75 con talla diana en p10) |
| Fenotipo | Cara redondeada | Dismorfias faciales leves | Cara redondeada, asimetrías faciales, epicantus, hendiduras palpebrales mongoloides, hipertelorismo, raíz nasal ancha, micrognatia, hombros estrechos | Cara redondeada, ptosis palpebral, epicantus, hendiduras palpebrales mongoloides, microcefalia, cabello de implantación baja, alteraciones dentales, prognatismo, hombros estrechos, micropene |
| Malformaciones congénitas | Clinodactilia, hernia inguinal, paladar hendido | Clinodactilia, sinóstosis radioulnar, problemas dentales | Sinóstosis radioulnar, displasia de cadera, malformaciones genitourinarias, paladar hendido, hernia inguinal, pies zambos, alteraciones cardíacas | Sinóstosis radioulnar, pies planos |
| Hipogonadismo hipergonadotropo | Sí | Sí | Sí | No valorable |
| Desarrollo y cognición | Retraso motor y del lenguaje (40-75%); CI medio de 89-102 | Temblores, retraso psicomotor; CI medio de 70-80 | Retraso motor y del lenguaje (100%); CI medio de 20-60 | Retraso motor y del lenguaje; CI<50 |
CI: cociente intelectual.
En conclusión, el cariotipo 48,XXXY/49,XXXXY es una aneuploidía infrecuente de los cromosomas sexuales que, pese a presentar alteraciones comunes al SK, se caracteriza por una mayor frecuencia de anomalías congénitas, especialmente sinóstosis radioulnar proximal, además de anomalías neuropsicológicas, en general, más graves que la forma 47,XXY. Se requiere seguimiento multidisciplinar de estos pacientes.
