





La neurofibromatosis tipo 1 (NF1) es la enfermedad neurocutánea más frecuente, pero el número de trabajos en que se recogen los datos clínicos y genéticos de un número amplio de niños es escaso.
Material y métodosSe recogen los datos clínicos, epidemiológicos, radiológicos y genéticos disponibles de 239 niños con NF1, atendidos en la consulta monográfica de NF1 entre enero del 2011 y diciembre del 2013.
ResultadosDoscientos treinta y nueve pacientes tenían un diagnóstico clínico y/o genético de NF1. La edad media al diagnóstico fue de 2,65±2,85 años. Cumplían los siguientes criterios diagnósticos: 99,6% manchas café con leche; 93,7% efélides axilares e inguinales; 7,1% lesión ósea característica; 38,1% neurofibromas, un 23% presentaron neurofibromas plexiformes; 31,4% glioma de vía óptica; 43,1% nódulos de Lisch, y un 28% tenían un familiar de primer grado afecto de NF1. En 86 pacientes se realizó el estudio genético de NF1. Se describen las mutaciones encontradas en 72 pacientes. Además, se detallan otros datos clínicos, que, ya por su frecuencia, ya por su gravedad, han sido asociados a NF1.
ConclusionesLa dificultad del diagnóstico clínico de la NF1 en edades precoces sigue siendo patente. A pesar de que se discute la necesidad o no de estudios complementarios en pacientes asintomáticos, la resonancia magnética craneal en niños con NF1 puede ser de gran ayuda en el diagnóstico clínico dada la alta incidencia del glioma de vía óptica que observamos en nuestra serie.
Neurofibromatosis type 1 (NF1) is the most common neurocutaneous disease, nevertheless the number of publications providing clinical and genetic data from a significant number of children is limited.
Material and methodsThe available clinical, epidemiological, radiological and genetic data from 239 children with NF1, who attended at a specialist NF1 clinic between January 2011 and December 2013 were recorded.
ResultsAll the 239 patients had a clinical and/or genetic diagnosis of NF1. The mean age at diagnosis was 2.65±2.85 years. In our series 99.6% met the diagnostic criteria of café au lait spots, 93.7% those of axillary and inguinal freckling, 7.1% showed typical bone lesion, 38.1% neurofibromas, 23% plexiform neurofibromas, 31.4% optic pathway glioma, Lisch nodules were present in 43.1%, and 28% patients had a first degree relative affected with NF1. The NF1 genetic study was performed in 86 patients, and a description of the gene mutations found in 72 of them is presented. Furthermore, other clinical data previously associated with NF1, either because of their frequency or their severity, are detailed.
ConclusionsThe difficulty for clinical diagnosis of NF1 early ages is still evident. Although, the need for further studies in asymptomatic patients is discussed, cranial MRI in children with NF1 may be helpful in the clinical diagnosis, given the high frequency of optic glioma observed in this cohort.
La neurofibromatosis tipo 1 (NF1) o enfermedad de von Recklinghausen es un trastorno genético con afectación predominante de la piel y el sistema nervioso, caracterizado por la presencia de manchas café con leche (MCCL), efélides axilares e inguinales, nódulos de Lisch en el iris, neurofibromas cutáneos y mayor riesgo de desarrollo tumoral. Esta enfermedad tiene una gran variabilidad clínica, incluso dentro de la misma familia y la morbimortalidad está asociada a sus complicaciones multisistémicas1,2. A pesar de ser la enfermedad neurocutánea más frecuente, el número de trabajos en que se recogen los datos clínicos y genéticos de un número amplio de niños con NF1 es escaso. La mayoría de las revisiones extraen los datos de los mismos estudios, principalmente de los trabajos realizados por Huson en el sudeste de Gales en 1988 y en 1989, pero estos trabajos incluyen un escaso porcentaje de población infantil3-5. El estudio más amplio centrado en población infantil que hemos encontrado en la literatura es el trabajo de Boulanger y Larbrisseau6, realizado en 2005. Los progresos en el conocimiento de su base genética han sido relevantes. Sin embargo, los estudios genéticos poseen escasos datos clínicos y los estudios fenotípicos, la mayoría antiguos, no incluyen datos genéticos. Nuestro objetivo es realizar una descripción exhaustiva del fenotipo en una amplia muestra de pacientes en edad pediátrica con NF1, aportando los estudios moleculares disponibles en los mismos.
Material y métodosEl presente trabajo consiste en un estudio descriptivo, transversal, observacional y retrospectivo. Se recogen los datos clínicos, epidemiológicos, radiológicos y genéticos disponibles de todos los pacientes de 0 a 18 años atendidos en la consulta monográfica de Neurofibromatosis entre enero del 2011 y diciembre del 2013 con los criterios clínicos diagnósticos de NF1 establecidos por el Instituto Nacional de Salud de EE. UU.7 (tabla 1) o con la confirmación genética de NF1. Para el diagnóstico de la NF Noonan (NFNS) deben cumplir además los criterios clínicos de síndrome de Noonan propuestos por van der Burgt8 (tabla 2).
Criterios clínicos diagnósticos de NF1
| Criterios diagnósticos de NF1: si cumplen al menos 2 de los siguientes |
|---|
| Al menos 6 manchas café con leche de diámetro>5mm en prepúberes y > 15mm en púberes |
| Presencia de moteado axilar y/o inguinal (efélides) |
| Al menos 2 neurofibromas o bien un neurofibroma plexiforme |
| Glioma de vía óptica |
| Lesión ósea sugerente de NF1: displasia de esfenoides, displasia o adelgazamiento de la cortical de huesos largos con o sin seudoartrosis |
| Dos o más nódulos de Lisch |
| Un familiar de primer grado con NF1 definida |
Modificado de NIH consensus development conference 19887.
Diagnóstico de síndrome de Noonan. Criterios (propuestos por van der Burgt): si cumplen una facies típica y un criterio mayor o 2 menores, o bien una facies sugestiva con 2 criterios mayores o 3 menores
| Criterios Diagnósticos | Mayores | Menores |
|---|---|---|
| Facies | Típica | Sugestiva |
| Cardíacos | Estenosis valvular pulmonarCardiomiopatía hipertrófica obstructivaElectrocardiograma típico | Otros defectos cardíacos |
| Talla | Percentil<3 | Percentil<10 |
| Pared del tórax | Pectus carinatum/excavatum | Tórax ancho |
| Historia Familiar | Familiar de primer grado con Síndrome de Noonan | Familiar de primer grado con diagnóstico sugerente de Síndrome de Noonan |
| Otros:- Retraso mental- Criptorquidia- Displasia linfática | Presencia de las 3 anomalías | Presencia de alguna de las 3 anomalías |
Modificado de van der Burgt8.
Se analizaron los datos de 332 pacientes, cumpliendo criterios de inclusión 239 niños procedentes de 225 familias; 14 niños tenían algún hermano o primo incluido en la muestra. En 67 pacientes la madre o el padre tenían una NF1 diagnosticada. Su edad media era 8,50±4,43 años (de 3 meses a 17 años) y la distribución por sexos paritaria. La edad media al diagnóstico fue de 2,65±2,85 años. El 91,8% fue diagnosticado antes de los 8 años. Un 2% cumplía 6 criterios clínicos diagnósticos, un 11% cumplía 5 criterios, un 34% cumplía 4 criterios, un 30% cumplía 3 criterios, un 22% cumplía 2 criterios. Dos pacientes (1%) cumplen un único criterio. Se trata de un niño de 5 años que cumple el criterio de MCCL y tiene diagnóstico genético de NF1, y otro niño con 9 años con antecedentes familiares (padre con NF1) y diagnóstico genético, que aunque presentaba MCCL no en número suficiente para cumplir el criterio de MCCL.
La edad media de cada subgrupo aumentaba conforme al número de criterios. Los criterios diagnósticos más frecuentemente observados fueron los cutáneos (MCCL y efélides axilares y/o inguinales) (fig. 1). En la tabla 3 se detallan además otros datos clínicos, que, ya por su frecuencia, ya por su gravedad, han sido asociados a NF1 y su frecuencia de aparición en esta serie. La edad de diagnóstico del glioma de vía óptica (GVO) está recogida en 70 pacientes con una edad media de 3,80±2,44 años. Solo 9 pacientes con GVO presentaron sintomatología grave (12%); los 9 cursaron con pérdida de visión. Hubo un total de 72 neurofibromas plexiformes (NFP), 23 internos y 49 externos, en 55 pacientes (ya que muchos presentaban más de un NFP). Un 30% de los NFP presentaron síntomas, 2 de mediastino con clínica respiratoria y escoliosis, 4 paraespinales o de raíces con dolor y escoliosis, 7 faciales con ptosis, exoftalmos o deformidad facial, y 9 localizados en el tronco o las extremidades produciendo deformidad y 5 de estos últimos además cursaron con dolor e impotencia funcional. En 87 pacientes se dispone de test psicométrico, siendo la media del cociente intelectual de 92,39±16,65. La edad de inicio de la marcha se recogió en 151 niños, siendo la edad media 15±3 meses. Los pacientes con cefalea secundaria fueron 3, en uno secundaria a hemorragia por síndrome de moyamoya y en los otros 2 secundaria a hidrocefalia. Cuatro pacientes presentaron afectación orbitotemporal con alteración ósea, de tejidos blandos y visual unilateral. Además, 2 de ellos asociaron GVO, 2 displasia de esfenoides, 3 neurofibroma plexiforme y 3 exoftalmos y ptosis. Se realizó resonancia magnética (RM) craneal a 199 pacientes; el 89,9% mostraba hiperseñales en secuencias T2. Su localización predominante fue en cerebelo, troncoencéfalo, ganglios basales, tálamo e hipocampo.
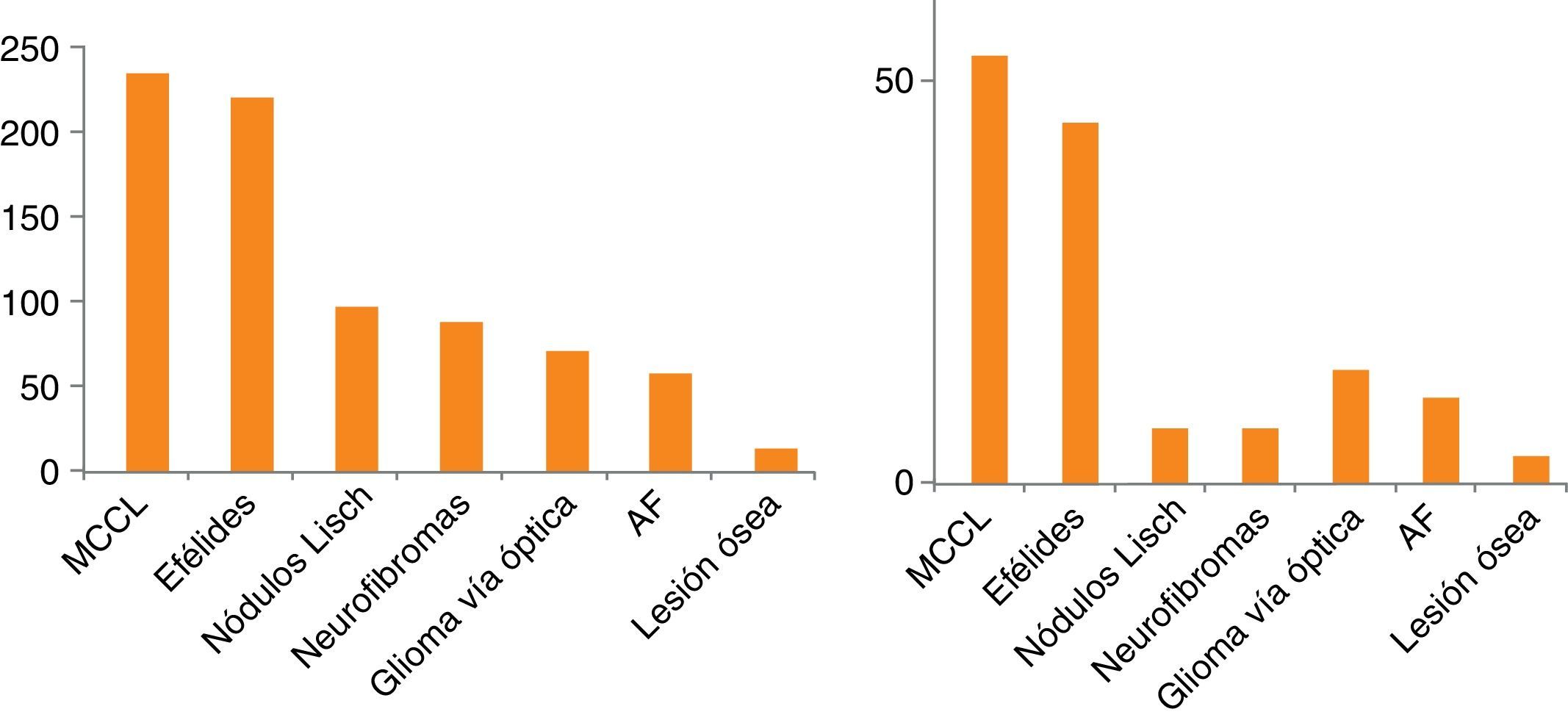
Síntomas y signos clínicos de los 239 niños con NF1
| Síntomas y signos asociados | n | % | Características |
|---|---|---|---|
| Criterio de manchas café con leche | 238 | 99,6 | |
| Criterio de efélides | 224 | 93,7 | |
| Criterio de neurofibromas | 91 | 38,1 | |
| Neurofibroma plexiforme | 55 | 23 | (Un total de 70 NFP en 55 pacientes)Localización: 9 facies,2 mucosas,5 extremidades inferiores,8 tronco,18 paraespinal y/o raíces,13 NFP cutáneos superficiales |
| NFP interno | 23 | 9,6 | |
| Criterio de nódulos de Lisch | 103 | 43,1 | |
| Criterio de glioma vía óptica | 75 | 31,4 | Localización principal: 30 unilateral,14 bilateral,23 quiasma,8 quiasma y cintillas ópticas |
| Criterio de lesión ósea (Displasia) | 17 | 7,1 | Localización: 11 en tibia,9 en fémur,uno en cúbito y radio,3 en esfenoides |
| Seudoartrosis | 9 | 3,7 | Localización: 8 en tibia,uno en cúbito y radio |
| Criterio de antecedentes familiares de NF1 | 67 | 28 | Familiar afectado: 36 la madre,29 el padre |
| Xantogranulomas | 15 | 6,3 | |
| Nevus anémico | 61 | 25,5 | |
| Hipertensión arterial | 5 | 2 | Etiología: una enfermedad de Kawasaki,una arteriopatía vasculorrenal,una secundaria a metilfenidato,2 desconocida (autolimitada) |
| Cardiopatía | 5 | 2 | Tipo: 3 estenosis de la válvula pulmonaruna insuficiencia de la válvula mitral,una comunicación interauricular |
| Síndrome de moyamoya | 4 | 1,6 | Clínica: 3 asintomáticosuno sintomático (hemorragia) |
| Dificultades de aprendizaje | 104 | 43,5 | 61 chicos+43 chicas |
| Retraso mental | 7 | 2,9 | Tipo: 6 leve+uno moderado |
| Trastorno de espectro autista | 1 | 0,41 | |
| Trastorno por déficit de atención e hiperactividad | 55 | 23 | Tipo: 22 inatento,32 combinadoUno hiperactivo/impulsivo |
| Malformaciones del sistema nervioso central | 10 | 4,1 | Etiología: 4 vasculopatías (moyamoya),un mielomeningocele,un disrafismo espinal2 estenosis del acueducto deSilvio con hidrocefalia,una hidrocefalia aislada,un Arnold Chiari tipo 1 |
| Retraso motor (hipotonía y torpeza) | 27 | 11,2 | |
| Diparesia espástica | 2 | 0,83 | Etiología: un mielomeningocele,un tumor medular |
| Crisis | 11 | 4,6 | |
| Epilepsia | 9 | 3,7 | Tipos: 5 epilepsia focal,2 epilepsia generalizada,una epilepsia ausencia,un síndrome de West |
| Cefalea | 41 | 17,1 | Tipos: 29 tensional,2 migraña,7 inespecífica,3 secundaria |
| Hiperseñales en secuencias T2 de la RM craneal | 179 | 74,8 | Localización 150 supra e infratentorial,20 infratentorial,9 supratentorial |
| Tumor glial de bajo grado | 95 | 39,7 | Localización: 75 glioma vía óptica,7 supratentoriales,8 tronco cerebral,5 medulares |
| Tumores malignos | 4 | 1,6 | Tipo: 1 neuroblastoma suprarrenal,3 rabdomiosarcomas |
| Escoliosis | 25 | 10,4 | Etiología: 6 de secundarias a NFP internoTipo: 13 leve4 moderada8 severa |
| Dismetría de extremidades inferiores | 8 | 3,3 | Etiología: 3 secundarias a NFP |
| Pubertad Precoz | 12 | 5 | Etiología: 7 secundarias a un glioma de vía óptica |
| Macrocefalia | 40 | 17 | |
| Talla baja (percentil<10) | 46 | 19 | |
| Talla baja (percentil<3) | 24 | 10 | |
| Neurofibromatosis Noonan | 12 | 5 | |
| Neurofibromatosis orbitotemporal | 4 | 1,6 |
n: número de pacientes; NFP: neurofibroma plexiforme.
En 86 pacientes se realizó el estudio genético de NF1. En 11 pacientes se realizó un estudio indirecto por haplotipos. En 75 pacientes se realizó un estudio directo mediante un cribado mutacional de ADNc NF1 con técnicas de ARN (cDNA-DHPLC denaturing high performance liquid chromatography) combinadas con técnicas basadas en MLPA (multiplex ligation-dependent probe amplification). Este método logra una sensibilidad del 95%9. Se encontró una mutación en 72 pacientes (de 67 familias) (tabla 4), siendo la mutación más frecuente tipo frameshift (mutación por desplazamiento del marco de lectura) (34,3%), seguido de nonsense (sin sentido) (22,3%). El síndrome de microdeleción se presentó en 5 pacientes, de los cuales 4 tenían dificultades generalizadas del aprendizaje y trastorno por déficit de atención (TDAH). Además, un paciente asociaba retraso mental moderado y otros 2, inteligencia límite. Tres pacientes tenían neurofibromas subcutáneos y uno presentaba además neurofibromas plexiformes internos. De los 12 pacientes con NFNS, en 9 se realizó estudio molecular que confirmó el diagnóstico de NF. Uno de ellos mediante un estudio indirecto y el resto eran los pacientes 8, 20, 28, 33, 36, 38, 55 y 64 de la tabla 4.
Análisis de las mutaciones encontradas en el gen NF1
| Caso | Exón | Mutación | mARN | Proteína | Clase | NT | Literatura |
|---|---|---|---|---|---|---|---|
| 1 | E36 | c.6657_6658dupTA | r.6657_6658dupua | p.Asn2220Ilefs*25 | FS | I | Nueva |
| 2 | E23-1 | c.3925_3928dupATCA | r.3925_3928dupauca | p.Thr1310Asnfs*5 | FS | I | Nueva |
| 3 | I28 | c.5205+5G>A | r.5152_5205del | p.Phe1719_Val1736del | SP | S | Descrita |
| 4,5,6 | E46 | c.7996_7997delAG | r.7996_7997delag | p.Ser2666Cysfs*5 | FS | D | Nueva |
| 7 | E30 | c.5613A>C | r.5613a>c | p.Leu1871Phe | MS | S | Nueva |
| 8 | I02 | c.205-1G>C | r.205_206delag | p.Arg69Asnfs*7 | SP | S | Nueva |
| 9 | E12a | c.1748A>G | r.1722_1748del | p.[Lys583Arg,Ser574_Lys583delinsArg] | SP/MS | S | Nueva |
| 10 | E34 | c.6513T>A | r.6513u>a | p.Tyr2171* | NS | S | Descrita |
| 11 | E04a | c.304_307delATGA | r.304_307delauga | p.Met102Aspfs*2 | FS | D | Nueva |
| 12 | I02 | c.204+1G>A | r.100_204del | p.Val34_Met68del | SP | S | Descrita |
| 13 | E23-1 | c.3938_3941delATTG | r.3938_3941delauug | p.Asp1313Glyfs*13 | FS | D | Nueva |
| 14 | I33 | c.6364+1delG | r.6085_6364del | p.Val2029Lysfs*7 | SP | D | Nueva |
| 15 | E22 | c.3835delA | r.3835dela | p.Ser1279Alafs*6 | FS | D | Nueva |
| 16 | E39 | c.7096_7101delAACTTT | p.Asn2366_Phe2367del | FS | D | Descrita | |
| 17 | E10c | c.1540C>T | r.1540c>u | p.Gln514* | NS | S | Nueva |
| 18 | E23-2 | c.4084C>T | r.4084c>u | p.Arg1362* | NS | S | Descrita |
| 19 | Deleción tipo i | r.0 | p.0 | DC | R | Descrita | |
| 20 | Deleción atípica | r.0 | p.0 | DC | R | Nueva | |
| 21 | E11 | c.1649T>C | r.1649u>c | p.Leu550Pro | MS | S | Nueva |
| 22 | E22 | c.3826C>T | r.3826c>u | p.Arg1276* | NS | S | Descrita |
| 23 | E23-2 | c.4071_4101del31 | r.4071_4101del31 | p.Pro1358Tyrfs*17 | FS | D | Nueva |
| 24 | E19b | c.3251delC | r.3251delc | p.Pro1068Leufs*12 | FS | D | Nueva |
| 25 | E13 | c.2033_2034insC | r.2033_2034insc | p.Ile679Aspfs*21 | FS | I | Descrita |
| 26 | E27a | c.4572C>G | r.4572c>g | p.Tyr1524* | NS | S | Nueva |
| 27 | I35 | c.6642-2A>G | r.6642_6757 | p.Phe2215Hisfs*7 | SP | S | Nueva |
| 28,29 | E29 | c.5425 C>G | r.5425 c>g | p.Arg1809Gly | MS | S | Nueva |
| 30 | E42 | c.7411C>T | r.7411c>u | p.Gln2471* | NS | S | Descrita |
| 31,32 | E12a_E27b | c.1722-?_4772+?del | r.[1642_4772del,1722_4772del] | p.[Ala548Valfs*9,Ser574Argfs*1229] | MD | R | Nueva |
| 33,34 | E29 | c.5425C>T | r.5425c>u | p.Arg1809Cys | MS | S | Descrita |
| 35 | E14 | c.2314insA | r.2314insA | p.Gly772Argfs*4 | FS | I | Nueva |
| 36 | E18 | c.3047G>A | r.3047g>a | p.Cys1016Tyr | MS | S | Nueva |
| 37 | E05 | c.699delA | r.699dela | p.Lys233Asnfs*47 | FS | D | Nueva |
| 38 | E05 | c.667T>C | r.667u>c | p.Trp223Arg | MS | S | Descrita |
| 39 | I31 | c.5943+1G>A | r.5901_5943del | p.Met1967Ilefs*10 | SP | S | Descrita |
| 40 | I01 | c.60+2T>G | r.o? | p.0? | SP | S | Nueva |
| 41 | E28 | c.5050delA | r.5050dela | p.Arg1684Glyfs*5 | FS | D | Nueva |
| 42 | E20 | c.3456_3457insAA | r.3456_3457insaa | p.Leu1153Asnfs*6 | FS | I | Nueva |
| 43 | E29 | c.5242C>T | r.5242c>u | p.Arg1748* | NS | S | Descrita |
| 44 | I21 | c.3709-1G>A | r.3709delg | p.Asp1237Metfs*3 | SP | S | Nueva |
| 45 | E21 | c.3553A>T | r.3553a>u | p.Lys1185* | NS | S | Nueva |
| 46 | E27a | c.4537C>T | r.4537c>u | p.Arg1513* | NS | S | Descrita |
| 47 | I23-1 | c.3974+260T>G | r.[3974_3975ins3974+1_3974+259, 3974_3975ins3974+180_3974+259, 3974_3975ins3974+1_3974+259 | p.[Leu1326Serfs*21, Leu1326Tyrfs*28, Leu1326Phefs*22] | SP | S | Nueva |
| 48 | E21 | c.[3572C>A;=] | r.3572c>a | p.Thr1191Lys | MS | S | Nueva |
| 49 | E28 | c.5104C>T | r.5104c>u | p.Gln1702* | NS | S | Nueva |
| 50 | E01 | c.55G>T | r.55g>u | p.Glu19* | NS | S | Descrita |
| 51 | E20 | c.3457_3460delCTCA | r.3457_3460delcuca | p.Leu1153Metfs*4 | FS | D | Descrita |
| 52 | E17 | c.2970_2971delAA | r.2970_2971delaa | p.Met991Aspfs*29 | FS | D | Descrita |
| 53 | Deleción tipo I | r.0 | p.0 | DC | R | Descrita | |
| 54 | E19b | c.3309_3312delTCTT | r3309_3312delucuu | p.Phe1103Leufs*8 | FS | D | Nueva |
| 55 | E32 | c.6056C>T | r.6056c>u | p.Ser2019Phe | MS | S | Nueva |
| 56 | E29 | c.5343dupC | r.5343dupc | p.Ile1782Hisfs*16 | FS | I | Nueva |
| 57 | E12a | c.1845G>T | r.1642_1845del | p.Ala548_Lys615del | SP | S | Descrita |
| 58 | E37 | c.6763delG | r.[6763delg,6758_6858del] | p.[Glu2255Argfs*4,Ala2253_Lys2286del] | SP | D | Nueva |
| 59 | E22 | c.3826C>T | r.3826c>u | p.Arg1276* | NS | S | Descrita |
| 60 | E16 | c.2657dup | r.2657dup | p.Asn886Lysfs*20 | FS | I | Nueva |
| 61 | I02 | c.205-1G>C | r.206_207del | p.Arg69Asnfs*7 | SP | S | Nueva |
| 62 | E27a | c.4469T>C | r.? | p.Leu1490Pro | MS | S | Nueva |
| 63 | E24 | c.4265C>A | r.4265c>a | p.Ser1422* | NS | S | Descrita |
| 64 | E10c | c.1540C>T | r.1540c>u | p.Gln514* | NS | S | Nueva |
| 65 | Deleción tipo I | r.0 | p.0 | DC | R | Descrita | |
| 66 | E21 | c.3548T>G | r.3548u>g | p.Leu1183Arg | MS | S | Nueva |
| 67 | E9 | c.1246C>T | r.1246c>u | p.Arg416* | NS | S | Descrita |
| 68 | E24 | c.4226dupA | r.4226dupa | p.Pro1410Alafs*4 | FS | I | Nueva |
| 69 | E19b | c.3309_3312delTCTT | r3309_3312delucuu | p.Phe1103Leufs*8 | FS | D | Nueva |
| 70 | E45 | c.7902delT | r.7902delu | p.Pro2634Profs*24 | FS | D | Nueva |
| 71 | E18 | c.3042delA | r.3042dela | p.Lys1014Asnfs*5 | FS | D | Nueva |
| 72 | Deleción tipo I | r.0 | p.0 | DC | R | Descrita |
D: deleción; DC: deleción completa del gen; FS: frameshift (cambio en la pauta de lectura); I: inserción; MD: deleción multiexón; MS: missense (cambio de aminoácido); NS: nonsense (mutación puntual que produce un codón de parada); NT: cambio nucleotídico; R: reordenamiento; S: sustitución; SP: splicing (mutación que afecta al correcto procesamiento del mARN).
Los resultados del presente trabajo son coherentes con los encontrados por otros autores3,6,10,11 en cuanto a la frecuencia de cada uno de los criterios clínicos (tabla 5). Una diferencia del presente estudio es que encontramos una prevalencia mucho mayor del criterio de GVO, que probablemente es debida al mayor número de RM craneales realizada en nuestro centro, donde se suele realizar esta prueba en aquellos pacientes asintomáticos mayores de 2 años de edad con criterios cutáneos de NF. Actualmente, no existe consenso respecto a las exploraciones complementarias a realizar en el diagnóstico y seguimiento de los niños asintomáticos con NF1, particularmente en lo referente a la indicación de la RM craneal. Pero dado que el diagnóstico precoz de las complicaciones es importante y la exploración neurológica y oftalmológica está limitada por la colaboración del paciente y esta, a su vez, determinada por la edad y presencia o no de retraso mental, hay autores que defienden la realización de la misma aun en ausencia de síntomas12-14, mientras otros la consideran indicada solo cuando los exámenes oculares no se pueden obtener de forma fiable15. Además, en numerosas ocasiones, la RM craneal resulta muy útil cómo herramienta de diagnóstico de la enfermedad6 (p. ej., un niño con MCCL asintomático en el que se encuentre un glioma óptico en la RM craneal). Por otro lado, las hiperseñales en las secuencias T2, tan frecuentes en la RM craneal, aunque no son diagnósticas ni patognomónicas, pueden apoyar la sospecha clínica de la NF1 en los niños pequeños, si estas se pueden definir con precisión y se presentan en zonas típicas, como el cerebelo, el tronco cerebral y los ganglios basales16. La RM craneal también permite el diagnóstico de complicaciones relacionadas con esta enfermedad. Principalmente, resulta de utilidad para el diagnóstico y el seguimiento de los GVO o de otra localización, pero también para otras complicaciones no tan frecuentes, como la hidrocefalia o la vasculopatía. Es justamente en las revisiones de NF1 realizadas por alguna patología neurológica en concreto (p. ej., en vasculopatía cerebral o epilepsia) donde algunos autores de manera directa o indirecta proponen detectar casos de patología neurológica aún silente13,14,17,18. Cuando analizamos solo los criterios clínicos diagnósticos en los pacientes de nuestra muestra con 6 años o menos (fig. 1), observamos cómo este criterio gana importancia en este grupo de edad dado que muchos niños aún no han desarrollado otros posibles criterios como la presencia de nódulos de Lisch o los neurofibromas cutáneos o subcutáneos. Esta idea se refuerza ante casos como los 2 pacientes que incluimos tras la confirmación genética de la enfermedad sin suficientes criterios como para establecer el diagnóstico de NF1, demostrando que el diagnóstico basado en criterios exclusivamente clínicos no siempre resulta sencillo durante los primeros años de vida.
Tabla comparativa entre diferentes cohortes de niños con neurofibromatosis tipo 1
| Síntomas y signos asociados | Estudio actual, 2014 | Boulanger y Larbrisseau62005 | Cnossen et al.101998 | Huson et al.31989 | Obringer et al.111989 |
|---|---|---|---|---|---|
| Número de pacientes | 239 | 279 | 150 | 168 | 160 |
| Manchas café con leche | 238 (99.6%) | 277 (99,3%) | 145 (96,7%) | 98,2% | 109 (97%) |
| Efélides | 224 (93,7%) | 59 (21,1%) | 128 (85,3%) | 70% | 91 (81%) |
| Neurofibromas | 49 (20%) | 107(38,4%) | 60 (49%) | 39,2% | 17 (15%) |
| Neurofibromas Plexiformes | 55 (23%) | 69 (24%) | 40 (26,6%) | 43,6% | |
| Neurofibrosarcomas | 0 (0%) | 5 (1,8%) | 3 (2%) | 0% | |
| Nódulos de Lisch | 103 (43,1%) | 137 (49,1%) | 78 (52%) | 83,9% | 34 (28%) |
| Glioma de vía óptica | 75 (31,4%) | 41 (14,7%) | 17 (11,3%) | 5,1% | 5 (4%) |
| Retraso mental(+ inteligencia límite) | 7 (2,9%)21 (8,7%) | 17 (6,1%) | 19 (17,2%) | ||
| Dificultades de aprendizaje | 104 (43%) | 85 (39%) | |||
| TDAH | 55 (23%) | 113 (40,5%) | |||
| Crisis | 11 (4,6%) | 9 (3,2%) | 1 (0,7%) | 5,1% | |
| Displasia Ósea | 17 (7,1%) | 20 (7,2%) | 11 (7,8%) | 5 (6%) | |
| Seudoartrosis | 9 (3,7%) | 10 (3,6%) | 3 (2%) | 5,1% | |
| Estenosis acueducto | 2 (0,8%) | 4 (1,4%) | 1 (0,7%) | 0% | |
| Pubertad precoz | 12 (5%) | 9 (3,2%) | 3 (2%) | ||
| Escoliosis | 25 (10,5%) | 33 (11,8%) | 2 | 5,1% | |
| Tumor cerebral | 15 (6,2%) | 4 (1,4%) | 4 (2,7%) | ||
| Síndrome de moyamoya | 4 (1,6%) | 5 (1,8%) | |||
| Hipertensión arterial | 5 (2%) | 4 (1,4%) | |||
| Macrocefalia (p>95) | 40 (16,7%) | 53 (19%) | |||
| Talla baja (p<10) | 46 (19,2%) | 50 (17,9%) | |||
| Hiperseñales en RM craneal en secuencias T2 | 179 (74%) | 81 (87,1%) |
p: percentil; RM: resonancia magnética; TDAH: trastorno por déficit de atención e hiperactividad.
Observamos una mayor frecuencia de neurofibromas plexiformes respecto el resto de neurofibromas, lo cual es probablemente debido a que se trata de una serie infantil, similar a lo reflejado en otros trabajos en edad pediátrica6. El subgrupo de neurofibromas plexiformes internos para aquellos que no se relacionan directamente con la piel19 parece particularmente útil en la clínica debido a que su presencia está subestimada al pasar inadvertidos a la exploración física, lo cual limita su conocimiento. La edad media con la que se detecta el GVO es de 3,8 años y resulta inferior a la referida en el trabajo de Boulanger y Larbrisseau6, de 5,1 años, lo cual puede estar en relación con la edad en que se realice la RM craneal. Sin embargo, el número de GVO que presentan sintomatología grave no es muy distinto del descrito por otros autores6. La presencia de displasias óseas y seudoartrosis es similar a la que encuentran en otros trabajos6,10, al igual que la preferencia por la localización en la tibia. Pero encontramos una frecuencia menor de displasia esfenoidal y ningún paciente con displasia vertebral. Los 4 pacientes con afectación orbitotemporal (diferenciada en la literatura como un subgrupo particular, NF orbitotemporal20) se presentaron como una entidad propia, agresiva y de etiología multifactorial.
Los nevus anémicos y el xantogranuloma juvenil recientemente están cobrando importancia clínica por su presencia en niños menores de 2 años, pues a esa edad los pacientes con NF de novo suelen cumplir un único criterio clínico, retrasándose así diagnóstico11,21. Ferrari et al.22 proponen como criterio menor el nevus anémico y el xantogranuloma, cuya asociación con NF1 es evidente y están descritos en edades precoces. Al ser un trabajo retrospectivo, suponemos que su frecuencia debe ser superior a la que referimos, dado que el nevus anémico debe buscarse a propósito durante la exploración frotando la zona afectada y el xantogranuloma juvenil tiende a desaparecer. Todos los pacientes tenían realizado control de presión arterial. A pesar de ello, no encontramos hipertensión arterial en la frecuencia descrita por otros autores seguramente con pacientes de más edad23. Otras enfermedades descritas en la NF1 de forma no incidental tampoco las hemos observado en nuestra casuística, seguramente por ser población infantil; es el caso del tumor glomus o de la esclerosis múltiple5.
En cuanto a las alteraciones cognitivas, es conocido que los pacientes con NF que presentan retraso mental son una minoría24. Nosotros encontramos un 2,9% de pacientes con retraso mental y hasta un 8,7% si incluimos inteligencia límite. En contraposición a diversos autores25 que han referido una alta prevalencia de trastorno del espectro autista en los niños con NF1, nosotros únicamente hallamos a un niño con el diagnóstico de trastorno de espectro autista. En la actualidad, se sabe que las publicaciones a este respecto son contradictorias y probablemente en mayor relación con el fenotipo cognitivo (retraso ejecutivo, alteraciones no verbales…) que con el social26.
Durante la infancia, suelen observarse signos neurológicos menores y discapacidad motriz con peor rendimiento en las tareas de evaluación de la motricidad fina, la coordinación motora, la destreza manual y el equilibrio27. No obstante, los estudios sobre el retraso motor en niños con NF1 son escasos, probablemente porque no se trata de un retraso severo, sino de la presencia de signos neurológicos menores, con una mayor dificultad para realizar una valoración objetiva. En nuestros pacientes, se refería un retraso motor en un 11%, pero al ser un estudio retrospectivo y tratarse en la mayoría de signos menores neurológicos puede no estar referido en las historias. La hipotonía, por otra parte, puede contribuir a la alteración motora, causando un retraso en la deambulación27. En la población general, la marcha liberada suele producirse entre los 13 y 15 meses de edad28. En el grupo con NF1, aunque dentro de la normalidad, la media está en el límite. No hemos encontrado en la literatura ningún estudio en que se refiera la edad de inicio de la marcha en un amplio grupo de niños con NF1.
Las malformaciones del sistema nervioso central, aunque son raras, no se consideran incidentales en esta patología. Nosotros, a pesar de que la mayoría de nuestros pacientes tenían realizada una RM craneal, no hemos observado una mayor incidencia de la comunicada previamente, a diferencia de lo que nos ha ocurrido con el GVO. Asimismo, todas las malformaciones que asociaban ya han sido descritas previamente en la NF15,18,29.
Consideramos importante destacar la presencia de cefalea secundaria a patología intracraneal en los niños con NF1 dada la frecuencia, la severidad de los casos y su implicación pronóstica y terapéutica. Observamos una frecuencia de epilepsia del 3,7% en nuestros pacientes, mayor que en la población general. Sin embargo, la frecuencia sigue siendo baja respecto de otros trastornos neurocutáneos14. En concordancia con la literatura, es más frecuente la epilepsia focal y que asocien déficit cognitivo30. En cuanto a la presencia de pubertad precoz (n=12), más de la mitad de los pacientes (7) presentaban un GVO que afectaba a la zona del quiasma, sin diferencias entre sexos, en contraposición a lo expuesto en la población general, en que la pubertad precoz por etiología orgánica tumoral es superior en el sexo masculino31. La talla baja (percentil<10) y la macrocefalia (percentil>95%) se presentaron con frecuencia similar, del 17 y el 19%, respectivamente. En ningún paciente se refería una etiología identificable, lo cual reafirma que probablemente se trate de una característica primaria32. En lo que se refiere a los tumores histológicamente malignos, es conocido que tienen una incidencia ligeramente mayor que en la población general. Nosotros observamos los tipos descritos de forma más frecuente en niños6. Ningún paciente presentó leucemia, incluidos los pacientes con antecedente de xantogranuloma juvenil. Esta asociación ya ha sido previamente cuestionada por otros autores19,22,33. La NF1 afecta a la vía enzimática de las RAS-quinasas, formando parte de los síndromes neurocardiofaciocutáneos o rasopatías, junto al síndrome de Noonan, el síndrome de Costello, el síndrome cardiofaciocutáneo, el síndrome de LEOPARD o el síndrome de Legius, entre otros, pudiendo compartir con estos un grado variable de características clínicas. Asimismo el recientemente descrito «síndrome de Legius», cuyo fenotipo clínico es similar a la NF1, ha planteado de nuevo que los criterios diagnósticos de NF1 deben ser revisados, dado que en la actualidad se consideran diagnóstico de NF1 el cumplir los criterios de MCCL y pecas axilares y o inguinales, y ambas manifestaciones pueden observarse en el síndrome de Legius. Entre el grado variable de características clínicas que comparten las rasopatías, se encuentra el dismorfismo facial34. Los pacientes con rasgos aislados o fenotipo indicativo de rasopatía en nuestra muestra eran numerosos. Pero solo hemos considerado NFNS a aquellos pacientes que cumplían criterios de síndrome de Noonan. En 9 de 12 pacientes se realizó estudio genético de NF1 confirmando la enfermedad. En conjunto, estos resultados orientan a la presencia de un fenotipo característico en algunos niños con NF1 y que comparten con otras rasopatías, y tal y como refieren de Luca et al., el tipo de NFNS podría tratarse de una variante de NF35. La variante NFNS está causada mayoritariamente por mutaciones en el gen NF1. De hecho, se propone la detección de mutaciones en el gen NF1 en aquellos pacientes con fenotipo Noonan y MCCL35.
En 83 pacientes el estudio genético confirmó la enfermedad. En 3 pacientes el análisis genético directo mutacional resultó negativo. Estos pacientes cumplían 2, 3 y 4 criterios, respectivamente. Se decidió no excluirlos del grupo, dado que cumplen criterios clínicos de NF1 y no superan el porcentaje de falsos negativos que presenta el estudio genético9,36. A pesar de que en otras series se ha referido hasta un 40% de mutaciones recurrentes, en pacientes no relacionados37, nosotros describimos 67 mutaciones (5 deleciones y 62 mutaciones) de 67 familias diferentes. Las mutaciones se distribuyen a lo largo de todo el gen NF1 y la mayoría son de tipo frameshift y nonsense, también halladas mayoritariamente por otros autores37-39. El síndrome de microdeleción se ha sido asociado a un fenotipo concreto10,40; en el caso de nuestros 5 pacientes, lo más llamativo eran las dificultades generalizadas del aprendizaje acompañadas de mayor o menor retraso, y los neurofibromas pero no encontramos malformaciones cerebrales estructurales, hipercrecimientos o malignización.
ConclusionesLas características fenotípicas, radiológicas y genéticas disponibles en 239 pacientes con NF1 en edad pediátrica presentadas conforman una imagen de los principales hallazgos y complicaciones que caracterizan a estos pacientes en una de las cohortes más numerosas en la literatura internacional. Con ellas se evidencia que la dificultad del diagnóstico clínico en edades precoces sigue siendo patente. El número de pacientes diagnosticados solo por criterios cutáneos, indistinguibles de un síndrome de Legius, es aún mayor en los niños, por ello indicamos que debería considerarse la posibilidad de unir los 2 criterios cutáneos diagnósticos (al menos 6 MCCL de diámetro superior a 5mm en prepúberes y superior a 15mm en los púberes y/o presencia de moteado axilar e inguinal) en un único criterio. A pesar de que se discute la necesidad o no de estudios complementarios en pacientes asintomáticos, la RM craneal en niños con NF1 puede ser de gran ayuda en el diagnóstico clínico dada la alta frecuencia del GVO que obtenemos en nuestra serie.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
