





Neurofibromatosis type 1 (NF1) is the most common neurocutaneous disease, nevertheless the number of publications providing clinical and genetic data from a significant number of children is limited.
Material and methodsThe available clinical, epidemiological, radiological and genetic data from 239 children with NF1, who attended at a specialist NF1 clinic between January 2011 and December 2013 were recorded.
ResultsAll the 239 patients had a clinical and/or genetic diagnosis of NF1. The mean age at diagnosis was 2.65±2.85 years. In our series 99.6% met the diagnostic criteria of café au lait spots, 93.7% those of axillary and inguinal freckling, 7.1% showed typical bone lesion, 38.1% neurofibromas, 23% plexiform neurofibromas, 31.4% optic pathway glioma, Lisch nodules were present in 43.1%, and 28% patients had a first degree relative affected with NF1. The NF1 genetic study was performed in 86 patients, and a description of the gene mutations found in 72 of them is presented. Furthermore, other clinical data previously associated with NF1, either because of their frequency or their severity, are detailed.
ConclusionsThe difficulty for clinical diagnosis of NF1 early ages is still evident. Although, the need for further studies in asymptomatic patients is discussed, cranial MRI in children with NF1 may be helpful in the clinical diagnosis, given the high frequency of optic glioma observed in this cohort.
La neurofibromatosis tipo 1 (NF1) es la enfermedad neurocutánea más frecuente, pero el número de trabajos en que se recogen los datos clínicos y genéticos de un número amplio de niños es escaso.
Material y métodosSe recogen los datos clínicos, epidemiológicos, radiológicos y genéticos disponibles de 239 niños con NF1, atendidos en la consulta monográfica de NF1 entre enero del 2011 y diciembre del 2013.
ResultadosDoscientos treinta y nueve pacientes tenían un diagnóstico clínico y/o genético de NF1. La edad media al diagnóstico fue de 2,65±2,85 años. Cumplían los siguientes criterios diagnósticos: 99,6% manchas café con leche; 93,7% efélides axilares e inguinales; 7,1% lesión ósea característica; 38,1% neurofibromas, un 23% presentaron neurofibromas plexiformes; 31,4% glioma de vía óptica; 43,1% nódulos de Lisch, y un 28% tenían un familiar de primer grado afecto de NF1. En 86 pacientes se realizó el estudio genético de NF1. Se describen las mutaciones encontradas en 72 pacientes. Además, se detallan otros datos clínicos, que, ya por su frecuencia, ya por su gravedad, han sido asociados a NF1.
ConclusionesLa dificultad del diagnóstico clínico de la NF1 en edades precoces sigue siendo patente. A pesar de que se discute la necesidad o no de estudios complementarios en pacientes asintomáticos, la resonancia magnética craneal en niños con NF1 puede ser de gran ayuda en el diagnóstico clínico dada la alta incidencia del glioma de vía óptica que observamos en nuestra serie.
Neurofibromatosis type 1 (NF1), also known as von Recklinghausen disease, is a genetic disorder that primarily involves the skin and the nervous system, characterised by the presence of café-au-lait macules (CALMs), axillary and inguinal freckling, iris Lisch nodules, cutaneous neurofibromas and a higher-than-average risk of tumour development. Its clinical expression is heterogeneous, even within a single family, and its morbidity and mortality are associated with multisystemic complications.1,2 Although it is the most prevalent neurocutaneous disorder, few studies have collected the clinical and genetic data of a broad number of children with NF1. Most reviews retrieve the data from the same studies, mainly from those conducted by Huson in south-east Wales in 1988 and 1989, but the proportion of children in these studies was low.3–5 The largest study focusing on the paediatric population that we found in the literature is the one conducted by Boulanger and Larbrisseau6 in 2005. There have been significant advances in the understanding of its genetic basis. However, genetic studies include few clinical data, and phenotypic studies, most of which were conducted long ago, do not include genetic data. Our aim was to provide an exhaustive phenotypical description in a broad sample of paediatric patients with NF1 along with all the available molecular data.
Materials and methodsWe conducted a descriptive, cross-sectional, observational retrospective study. We collected the clinical, epidemiological, radiological and genetic data available for every patient 0–18 years of age that received care at a specialist neurofibromatosis clinic between January 2011 and December 2013 and met the clinical diagnostic criteria for NF1 established by the National Institute of Health of the United States7 (Table 1) or with a genetic diagnosis of NF1. The diagnosis of neurofibromatosis-Noonan syndrome (NFNS) required that the patient also met the clinical criteria for Noonan syndrome proposed by van der Burgt8 (Table 2).
Clinical diagnostic criteria for NF1.
| Diagnostic criteria for NF1: meet at least 2 of the following |
| At least 6 café-au-lait macules with a diameter >5mm in prepubertal children and >15mm after puberty |
| Presence of axillary or inguinal spots (freckling) |
| At least 2 neurofibromas or one plexiform neurofibroma |
| Optic nerve glioma |
| Bone lesion suggestive of NF1: sphenoid dysplasia, long-bone dysplasia or cortical thinning with or without pseudarthrosis |
| Two or more Lisch nodules |
| First-degree relative with definite NF1 |
Diagnosis of Noonan syndrome. Criteria (proposed by van der Burgt): typical facies and one major criterion or 2 minor criteria, or suggestive facies with 2 major or 3 minor criteria.
| Diagnostic criteria | Major | Minor |
|---|---|---|
| Facial | Typical facies | Suggestive facies |
| Cardiac | Pulmonary valve stenosisHypertrophic obstructive cardiomyopathyTypical electrocardiogram | Other defect |
| Height | <3rd percentile | <10th percentile |
| Chest wall | Pectus carinatum/excavatum | Broad thorax |
| Family history | First degree relative with definite Noonan syndrome | First degree relative with suggestive Noonan syndrome |
| Other:- Mental retardation- Cryptorchidism- Lymphatic dysplasia | Presence of all 3 anomalies | Presence of one of the 3 anomalies |
We analysed the data of 332 patients, of which 239 children from 225 families met the inclusion criteria; 14 children had either siblings or cousins that were included in the sample. Sixty-seven patients had a father or mother diagnosed with NF1. The mean age of the patients was 8.50±4.43 years (range, 3 months–17 years) and the sex distribution was homogeneous. The mean age at diagnosis was 2.65±2.85 years. Children were diagnosed before age 8 years in 91.8% of the cases. Two percent met six clinical diagnostic criteria, 11% five criteria, 34% four criteria, 30% three criteria and 22% two criteria. Two patients (1%) met a single criterion. One was a 5-year-old boy that had CALMs and a genetic diagnosis of NF1, and the other a 9-year-old boy with a positive family history (father with NF1) and a genetic diagnosis that had CALMs but not in enough quantity to meet the CALM criterion.
The mean age of each subgroup increased with the number of criteria. The diagnostic criteria observed most frequently were cutaneous criteria (CALMs and axillary and/or inguinal freckles) (Fig. 1). Table 3 presents data for additional clinical features previously associated with NF1 due to their frequency or severity, along with their frequency in our series. The age at diagnosis of optic nerve glioma (ONG) was documented in 70 patients, with a mean age of 3.80±2.44 years. Only 9 patients with ONG (12%) had severe symptoms; all 9 had loss of vision. There were a total of 72 plexiform neurofibromas (PNFs), 23 of which were internal and 49 external, in 55 patients (as many had more than one PNF). Thirty percent of the PNFs were symptomatic: 2 were located in the mediastinum and were accompanied by respiratory symptoms and scoliosis, 4 were paraspinal or at the nerve roots and were associated with pain and scoliosis, 7 were facial with ptosis, exophthalmos or facial deformity, and 9 were located in the torso or limbs and caused deformities, with 5 of the latter manifesting with pain and functional impotence. A psychometric test was performed in 87 patients, the mean IQ of whom was 92.39±16.65. The age the patient started walking was documented in 151 children, and the mean age was 15±3 months. Three patients experienced secondary headache, which was due to moyamoya disease in one, and to hydrocephalus in the other two. Four patients had orbitotemporal involvement with unilateral bone, soft-tissue and vision abnormalities. In addition, two of them had ONG, two had sphenoid dysplasia, three had plexiform neurofibroma and three had exophthalmos and ptosis. Cranial magnetic resonance imaging (MRI) was performed in 199 patients, and hyperintensities were observed in 89.9% in the T2-weighted scan. Hyperintensities were most frequently located at the cerebellum, brainstem, basal ganglia, thalamus and hippocampus.

Clinical signs and symptoms of the 239 children with NF1.
| Associated signs and symptoms | n | % | Characteristics |
|---|---|---|---|
| Café-au-lait macules criterion | 238 | 99.6 | |
| Freckling criterion | 224 | 93.7 | |
| Neurofibromas criterion | 91 | 38.1 | |
| Plexiform neurofibroma | 55 | 23 | (A total of 70 PFN in 55 patients)Location: 9 face, 2 mucosa, 5 lower extremities, 8 torso, 18 paraspinal and/or nerve roots,13 superficial skin PFN |
| Internal PNF | 23 | 9.6 | |
| Lisch nodules criterion | 103 | 43.1 | |
| Optic nerve glioma criterion | 75 | 31.4 | Main location: 30 unilateral,14 bilateral,23 optic chiasm,8 optic chiasm and tracts |
| Bone lesion criterion (Dysplasia) | 17 | 7.1 | Location: 11 in the tibia,9 in the femur,1 in ulna and radius3 in sphenoid bone |
| Pseudarthrosis | 9 | 3.7 | Location: 8 in tibia,1 in ulna and radius |
| Family history of NF1 criteria | 67 | 28 | Affected relative: mother (36), father (29) |
| Xanthogranulomas | 15 | 6.3 | |
| Nevus anemicus | 61 | 25.5 | |
| Hypertension | 5 | 2 | Aetiology: 1 Kawasaki disease,1 renal artery disease,1 secondary to methylphenidate,2 unknown (self-limited) |
| Heart disease | 5 | 2 | Type: 3 pulmonary valve stenosis1 mitral insufficiency,1 atrial septal defect |
| Moyamoya disease | 4 | 1.6 | Manifestations: 3 asymptomatic1 symptomatic (haemorrhage) |
| Learning disabilities | 104 | 43.5 | 61 male+43 female |
| Intellectual disability | 7 | 2.9 | Type: 6 mild+1 moderate |
| Autism-spectrum disorders | 1 | 0.41 | |
| Attention deficit hyperactivity disorder | 55 | 23 | Type: 22 inattentive,32 combined1 hyperactive-impulsive |
| Central nervous system malformations | 10 | 4.1 | Aetiology: 4 vascular disease (moyamoya),1 myelomeningocele,1 spinal dysraphism2 aqueductal stenosis with hydrocephalus,1 isolated hydrocephalus,1 Arnold Chiari type 1 |
| Motor delay (hypotonia and clumsiness) | 27 | 11.2 | |
| Spastic diplegia | 2 | 0.83 | Aetiology: 1 myelomeningocele,1 spinal cord tumour |
| Seizures | 11 | 4.6 | |
| Epilepsy | 9 | 3.7 | Types: 5 focal epilepsy,2 generalised epilepsy,1 absence epilepsy1 West syndrome |
| Headache | 41 | 17.1 | Types: 29 tension,2 migraine,7 nonspecific,3 secondary |
| Hyperintensities in T2-weighted head MRI | 179 | 74.8 | Location: 150 supra- and infratentorial,20 infratentorial,9 supratentorial |
| Low grade glioma | 95 | 39.7 | Location: 75 optic nerve glioma,7 supratentorial,8 brainstem,5 spinal cord |
| Malignant tumours | 4 | 1.6 | Type: 1 suprarenal neuroblastoma,3 rhabdomyosarcomas |
| Scoliosis | 25 | 10.4 | Aetiology: 6 secondary to internal PFNType: 13 mild4 moderate8 severe |
| Leg length discrepancy | 8 | 3.3 | Aetiology: 3 secondary to PFN |
| Precocious puberty | 12 | 5 | Aetiology: 7 secondary to optic nerve glioma |
| Macrocephaly | 40 | 17 | |
| Short stature (<10th percentile) | 46 | 19 | |
| Short stature (<3rd percentile) | 24 | 10 | |
| Neurofibromatosis-Noonan syndrome | 12 | 5 | |
| Orbitotemporal neurofibromatosis | 4 | 1.6 |
n, number of patients; PNF, plexiform neurofibroma.
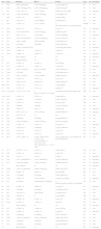
Genetic analysis of the NF1 gene was performed in 86 patients. Indirect haplotyping was performed in 11 patients. A direct study was performed in 75 patients by means of a mutation screening of the NF1 cDNA using RNA techniques (cDNA-denaturing high performance liquid chromatography [DHPLC]) combined with techniques based on multiplex ligation-dependent probe amplification (MLPA). This method has a sensitivity of 95%.9 Mutations were detected in 72 patients from 67 families (Table 4), with the most common type being frameshift mutations (mutations due to a shift in the reading frame, 34%), followed by nonsense mutation (22.3%). Five patients had microdeletion syndrome, of which four had general learning disabilities and attention deficit hyperactivity disorder (ADHD). Also, one patient had moderate intellectual disability and another 3 borderline intellectual functioning. Three patients had subcutaneous neurofibromas and one also had internal plexiform neurofibromas. Of the 12 patients with NFNS, 9 underwent genetic testing that confirmed the NF diagnosis. Confirmation was done by indirect testing in one, and the others correspond to patients 8, 20, 28, 33, 36, 38, 55 and 64 in Table 4.
Analysis of the mutations found in the NF1 gene.
| Case | Exon | Mutation | mRNA | Protein | Type | NC | Literature |
|---|---|---|---|---|---|---|---|
| 1 | E36 | c.6657_6658dupTA | r.6657_6658dupua | p.Asn2220Ilefs*25 | FS | I | New |
| 2 | E23-1 | c.3925_3928dupATCA | r.3925_3928dupauca | p.Thr1310Asnfs*5 | FS | I | New |
| 3 | I28 | c.5205+5G>A | r.5152_5205del | p.Phe1719_Val1736del | SP | S | Reported |
| 4,5,6 | E46 | c.7996_7997delAG | r.7996_7997delag | p.Ser2666Cysfs*5 | FS | D | New |
| 7 | E30 | c.5613A>C | r.5613a>c | p.Leu1871Phe | MS | S | New |
| 8 | I02 | c.205-1G>C | r.205_206delag | p.Arg69Asnfs*7 | SP | S | New |
| 9 | E12a | c.1748A>G | r.1722_1748del | p.[Lys583Arg,Ser574_Lys583delinsArg] | SP/MS | S | New |
| 10 | E34 | c.6513T>A | r.6513u>a | p.Tyr2171* | NS | S | Reported |
| 11 | E04a | c.304_307delATGA | r.304_307delauga | p.Met102Aspfs*2 | FS | D | New |
| 12 | I02 | c.204+1G>A | r.100_204del | p.Val34_Met68del | SP | S | Reported |
| 13 | E23-1 | c.3938_3941delATTG | r.3938_3941delauug | p.Asp1313Glyfs*13 | FS | D | New |
| 14 | I33 | c.6364+1delG | r.6085_6364del | p.Val2029Lysfs*7 | SP | D | New |
| 15 | E22 | c.3835delA | r.3835dela | p.Ser1279Alafs*6 | FS | D | New |
| 16 | E39 | c.7096_7101delAACTTT | p.Asn2366_Phe2367del | FS | D | Reported | |
| 17 | E10c | c.1540C>T | r.1540c>u | p.Gln514* | NS | S | New |
| 18 | E23-2 | c.4084C>T | r.4084c>u | p.Arg1362* | NS | S | Reported |
| 19 | Type I deletion | r.0 | p.0 | FD | R | Reported | |
| 20 | Atypical deletion | r.0 | p.0 | FD | R | New | |
| 21 | E11 | c.1649T>C | r.1649u>c | p.Leu550Pro | MS | S | New |
| 22 | E22 | c.3826C>T | r.3826c>u | p.Arg1276* | NS | S | Reported |
| 23 | E23-2 | c.4071_4101del31 | r.4071_4101del31 | p.Pro1358Tyrfs*17 | FS | D | New |
| 24 | E19b | c.3251delC | r.3251delc | p.Pro1068Leufs*12 | FS | D | New |
| 25 | E13 | c.2033_2034insC | r.2033_2034insc | p.Ile679Aspfs*21 | FS | I | Reported |
| 26 | E27a | c.4572C>G | r.4572c>g | p.Tyr1524* | NS | S | New |
| 27 | I35 | c.6642-2A>G | r.6642_6757 | p.Phe2215Hisfs*7 | SP | S | New |
| 28,29 | E29 | c.5425 C>G | r.5425 c>g | p.Arg1809Gly | MS | S | New |
| 30 | E42 | c.7411C>T | r.7411c>u | p.Gln2471* | NS | S | Reported |
| 31,32 | E12a_E27b | c.1722-?_4772+?del | r.[1642_4772del,1722_4772del] | p.[Ala548Valfs*9,Ser574Argfs*1229] | MD | R | New |
| 33,34 | E29 | c.5425C>T | r.5425c>u | p.Arg1809Cys | MS | S | Reported |
| 35 | E14 | c.2314insA | r.2314insA | p.Gly772Argfs*4 | FS | I | New |
| 36 | E18 | c.3047G>A | r.3047g>a | p.Cys1016Tyr | MS | S | New |
| 37 | E05 | c.699delA | r.699dela | p.Lys233Asnfs*47 | FS | D | New |
| 38 | E05 | c.667T>C | r.667u>c | p.Trp223Arg | MS | S | Reported |
| 39 | I31 | c.5943+1G>A | r.5901_5943del | p.Met1967Ilefs*10 | SP | S | Reported |
| 40 | I01 | c.60+2T>G | r.o? | p.0? | SP | S | New |
| 41 | E28 | c.5050delA | r.5050dela | p.Arg1684Glyfs*5 | FS | D | New |
| 42 | E20 | c.3456_3457insAA | r.3456_3457insaa | p.Leu1153Asnfs*6 | FS | I | New |
| 43 | E29 | c.5242C>T | r.5242c>u | p.Arg1748* | NS | S | Reported |
| 44 | I21 | c.3709-1G>A | r.3709delg | p.Asp1237Metfs*3 | SP | S | New |
| 45 | E21 | c.3553A>T | r.3553a>u | p.Lys1185* | NS | S | New |
| 46 | E27a | c.4537C>T | r.4537c>u | p.Arg1513* | NS | S | Reported |
| 47 | I23-1 | c.3974+260T>G | r.[3974_3975ins3974+1_3974+259, 3974_3975ins3974+180_3974+259, 3974_3975ins3974+1_3974+259] | p.[Leu1326Serfs*21, Leu1326Tyrfs*28, Leu1326Phefs*22] | SP | S | New |
| 48 | E21 | c.[3572C>A;=] | r.3572c>a | p.Thr1191Lys | MS | S | New |
| 49 | E28 | c.5104C>T | r.5104c>u | p.Gln1702* | NS | S | New |
| 50 | E01 | c.55G>T | r.55g>u | p.Glu19* | NS | S | Reported |
| 51 | E20 | c.3457_3460delCTCA | r.3457_3460delcuca | p.Leu1153Metfs*4 | FS | D | Reported |
| 52 | E17 | c.2970_2971delAA | r.2970_2971delaa | p.Met991Aspfs*29 | FS | D | Reported |
| 53 | Type I deletion | r.0 | p.0 | FD | R | Reported | |
| 54 | E19b | c.3309_3312delTCTT | r3309_3312delucuu | p.Phe1103Leufs*8 | FS | D | New |
| 55 | E32 | c.6056C>T | r.6056c>u | p.Ser2019Phe | MS | S | New |
| 56 | E29 | c.5343dupC | r.5343dupc | p.Ile1782Hisfs*16 | FS | I | New |
| 57 | E12a | c.1845G>T | r.1642_1845del | p.Ala548_Lys615del | SP | S | Reported |
| 58 | E37 | c.6763delG | r.[6763delg,6758_6858del] | p.[Glu2255Argfs*4,Ala2253_Lys2286del] | SP | D | New |
| 59 | E22 | c.3826C>T | r.3826c>u | p.Arg1276* | NS | S | Reported |
| 60 | E16 | c.2657dup | r.2657dup | p.Asn886Lysfs*20 | FS | I | New |
| 61 | I02 | c.205-1G>C | r.206_207del | p.Arg69Asnfs*7 | SP | S | New |
| 62 | E27a | c.4469T>C | r.? | p.Leu1490Pro | MS | S | New |
| 63 | E24 | c.4265C>A | r.4265c>a | p.Ser1422* | NS | S | Reported |
| 64 | E10c | c.1540C>T | r.1540c>u | p.Gln514* | NS | S | New |
| 65 | Type I deletion | r.0 | p.0 | FD | R | Reported | |
| 66 | E21 | c.3548T>G | r.3548u>g | p.Leu1183Arg | MS | S | New |
| 67 | E9 | c.1246C>T | r.1246c>u | p.Arg416* | NS | S | Reported |
| 68 | E24 | c.4226dupA | r.4226dupa | p.Pro1410Alafs*4 | FS | I | New |
| 69 | E19b | c.3309_3312delTCTT | r3309_3312delucuu | p.Phe1103Leufs*8 | FS | D | New |
| 70 | E45 | c.7902delT | r.7902delu | p.Pro2634Profs*24 | FS | D | New |
| 71 | E18 | c.3042delA | r.3042dela | p.Lys1014Asnfs*5 | FS | D | New |
| 72 | Type I deletion | r.0 | p.0 | FD | R | Reported |
D, deletion; FD, full gene deletion; FS, frameshift (change in the reading frame); I, insertion; MD, multi-exon deletion; MS, missense (amino acid change); NS: nonsense (point mutation that results in a stop codon); NC, nucleotide change; R, rearrangement; S, substitution; SP, splicing (mutation that affects the correct processing of mRNA).
The results of our study were consistent with those of other authors3,6,10,11 when it came to the frequency of each of the clinical criteria (Table 5). Our study did diverge in that we found a much higher prevalence of the ONG criterion, which was probably due to the greater number of cranial MRI scans performed in our hospital, where this test is usually done in asymptomatic patients older than 2 years that meet cutaneous criteria for NF. At present, there is no consensus on the diagnostic tests that should be performed in the diagnosis and followup of asymptomatic children with NF1, especially in relation to the indication of cranial MRI. But given the importance of diagnosing NF1 complications early and that adequate neurological and ophthalmological examinations depend on the cooperation of the patient, which in turn is determined by patient age and presence or absence of intellectual disability, some authors propose that MRI scans should be done even in asymptomatic patients,12–14 while others consider that it is only indicated when eye exams cannot be carried out appropriately.15 Also, a cranial MRI scan can often be of great help in the diagnosis of NF16 (for instance, in asymptomatic children with CALMs in whom optic nerve gliomas are detected by means of cranial MRI). On the other hand, although hyperintensities in T2-weighted images, so frequent in cranial MRI, are neither diagnostic nor pathognomonic, they can support the clinical suspicion of NF1 in young children if they are well defined and appear in typical regions such as the cerebellum, the brainstem and the basal ganglia.16 Cranial MRI also allows the diagnosis of complications associated with this disease. It is mostly useful to diagnose and monitor gliomas in the optic nerve or other locations, but also other complications that are less frequent, such as hydrocephalus or vascular diseases. It is precisely in studies that review the records of patients with NF1 with a focus on a specific neurologic pathology (such as cerebrovascular disease or epilepsy) that some authors have directly or indirectly proposed testing to detect cases of still-asymptomatic neurologic pathology.13,14,17,18 When we analysed the prevalence of the clinical diagnostic criteria in patients 6 years of age or younger in our sample (Fig. 1) we observed that this criterion is more important in this age group, as many children have yet to develop other potential criteria such as Lisch nodules or cutaneous or subcutaneous neurofibromas. This notion is supported by cases such as those of the two patients included in the study after the disease was confirmed by genetic testing that did not meet enough criteria for the diagnosis of NF1, demonstrating that it is not always easy to make the diagnosis based solely on clinical criteria in the early years of life.
Table comparing different cohorts of children with neurofibromatosis type 1.
| Associated signs and symptoms | Our study, 2014 | Boulanger and Larbrisseau62005 | Cnossen et al,101998 | Huson et al,31989 | Obringer et al,111989 |
|---|---|---|---|---|---|
| Number of patients | 239 | 279 | 150 | 168 | 160 |
| Café-au-lait macules | 238 (99.6%) | 277 (99.3%) | 145 (96.7%) | 98.2% | 109 (97%) |
| Freckling | 224 (93.7%) | 59 (21.1%) | 128 (85.3%) | 70% | 91 (81%) |
| Neurofibromas | 49 (20%) | 107 (38.4%) | 60 (49%) | 39.2% | 17 (15%) |
| Plexiform neurofibromas | 55 (23%) | 69 (24%) | 40 (26.6%) | 43.6% | |
| Neurofibrosarcomas | 0 (0%) | 5 (1.8%) | 3 (2%) | 0% | |
| Lisch nodules | 103 (43.1%) | 137 (49.1%) | 78 (52%) | 83.9% | 34 (28%) |
| Optic nerve glioma | 75 (31.4%) | 41 (14.7%) | 17 (11.3%) | 5.1% | 5 (4%) |
| Intellectual disability(+ borderline intellectual functioning) | 7 (2.9%)21 (8.7%) | 17 (6.1%) | 19 (17.2%) | ||
| Learning disabilities | 104 (43%) | 85 (39%) | |||
| ADHD | 55 (23%) | 113 (40.5%) | |||
| Seizures | 11 (4.6%) | 9 (3.2%) | 1 (0.7%) | 5.1% | |
| Bone dysplasia | 17 (7.1%) | 20 (7.2%) | 11 (7.8%) | 5 (6%) | |
| Pseudarthrosis | 9 (3.7%) | 10 (3.6%) | 3 (2%) | 5.1% | |
| Aqueductal stenosis | 2 (0.8%) | 4 (1.4%) | 1 (0.7%) | 0% | |
| Precocious puberty | 12 (5%) | 9 (3.2%) | 3 (2%) | ||
| Scoliosis | 25 (10.5%) | 33 (11.8%) | 2 | 5.1% | |
| Brain tumour | 15 (6.2%) | 4 (1.4%) | 4 (2.7%) | ||
| Moyamoya disease | 4 (1.6%) | 5 (1.8%) | |||
| Hypertension | 5 (2%) | 4 (1.4%) | |||
| Macrocephaly (>95‰) | 40 (16.7%) | 53 (19%) | |||
| Short stature (<10‰) | 46 (19.2%) | 50 (17.9%) | |||
| Hyperintensities in T2-weighted brain MRI T2 | 179 (74%) | 81 (87.1%) |
‰, percentile; ADHD, attention deficit hyperactivity disorder; MRI, magnetic resonance imaging.
We observed a greater frequency of plexiform neurofibromas compared to the other types. This was probably due to our study being conducted on the paediatric population, which is consistent with the findings of other studies on this age group.6 The subgroup of internal plexiform neurofibromas without a direct connection to the skin19 comprise a particularly relevant manifestation, as its prevalence is underestimated because they often go undetected in the physical examination, which limits our knowledge of them. The mean age at which ONG was detected was 3.8 years, which was lower than the age of 5.1 years reported by Boulanger and Larbrisseau,6 which could be related to the age at which head MRI scans were performed. However, the frequency of ONG with severe symptoms did not differ significantly from that described by other authors.6 The prevalence of bone dysplasia and pseudarthrosis was similar to the prevalence reported in other publications,6,10 as was the predominant localisation in the tibia. However, we found a lower prevalence of sphenoid dysplasia and no patient had vertebral dysplasia. The four patients with orbitotemporal involvement (set apart in the literature as a specific subgroup, orbitotemporal NF20) had a clinical presentation that appeared as a separate entity, aggressive and with a multifactorial aetiology.
Recently, the clinical relevance of nevus anemicus and juvenile xanthogranuloma has been growing due to their presence in children younger than 2 years, as at these ages patients with de novo NF usually meet a single clinical criterion, and this results in diagnostic delays.11,21 Ferrari et al22 propose adding nevus anemicus and xanthogranuloma as minor criteria, as they have a demonstrated association with NF1 and have been reported in young patients. Since this is a retrospective study, we suppose that their prevalence must be greater than the one we are reporting, as nevus anemicus must be purposefully sought by the clinician during the examination by rubbing the affected area, and juvenile xanthogranuloma tends to resolve spontaneously. All patients had been assessed for arterial blood pressure. However, hypertension was not found as frequently as it was by other authors that were probably studying older patients.23 There are other diseases described to have a non-incidental association with NF1, such as glomus tumours and multiple sclerosis,5 that we also did not find in our case series, probably because our study was conducted on the paediatric population.
As for cognitive disorders, it is known that only a minority of patients with NF present with intellectual disability.24 In our sample, we found intellectual disability in 2.9% of the patients, which rose to 8.7% if we included borderline intellectual functioning. Unlike what has been reported by several authors25 that found a high prevalence of autism spectrum disorders in children with NF1, we only found one child with such a diagnosis. At present, it is known that the literature on this subject is contradictory, and is probably related to the cognitive phenotype (features like deficits in executive function and nonverbal analysis) than with social features.26
Minor neurological signs and motor disability are usually observed in childhood, and patients perform more poorly in tasks used to evaluate fine motor skills, manual dexterity and balance.27 However, there are few studies on motor delay in children with NF1, probably because the delay is not severe, but consists mostly in the presence of minor neurological signs, so that it is difficult to perform an objective assessment. In our sample, motor delays were found in 11% of the patients, but considering the retrospective design of our study and that most of the neurologic signs were minor, they may not always have been documented in the medical records. On the other hand, hypotonia may contribute to motor abnormalities, causing delays in walking.27 In the general population, independent walking usually occurs at 13–15 months of age.28 The mean age in patients with NF1, while being within the normal range, is at the limit. We did not find any studies in the literature with a large sample of children with NF1 that reported the age the children learned to walk.
While malformations of the central nervous system rarely occur in this disease, they are not considered coincidental. Even though cranial MRI was performed in most of our patients, we did not observe a higher incidence of these malformations than that previously reported, unlike what occurred with ONG. Furthermore, all the documented malformations had been previously described in association with NF1.5,18,29
We ought to underscore the presence of headache secondary to intracranial pathology in children with NF1 given the frequency and severity of these cases and their prognostic and therapeutic implications. We observed a 3.7% prevalence of epilepsy in our patients, greater than that of the general population. However, this prevalence is still small compared to that in other neurocutaneous disorders.14 Consistent with the literature, the most frequent form was focal epilepsy associated with cognitive impairment.30 As for patients with precocious puberty (n=12), more than half (7) had an ONG that affected the optic chiasm, with no difference in prevalence between the sexes, unlike what has been described for the general population, in which organic precocious puberty caused by tumours is more prevalent in males.31 Short stature (<10th percentile) and macrocephaly (>95th percentile) had similar prevalences of 17% and 19%, respectively. No identifiable aetiology had been documented for any of the patients, which supports that this is probably a primary characteristic.32 When it comes to malignant tumours, it is known that their prevalence is slightly higher in patients with NF1 than in the general population. We observed the types most frequently reported in children.6 None of the patients had leukaemia, including those with a history of juvenile xanthogranuloma. This association has already been questioned by other authors.19,22,33 Neurofibromatosis type 1 affects the Ras kinase pathway, and is one of the neurocardiofaciocutaneous syndromes or RASopathies, along with Noonan syndrome, Costello syndrome, cardiofaciocutaneous syndrome, LEOPARD syndrome or Legius syndrome, among others, with which it shares a variable range of clinical characteristics. In addition, the recently described Legius syndrome, which has a clinical phenotype similar to that of NF1, presents the need to once more update the diagnostic criteria for NF1, as at present NF1 can be diagnosed if the patient meets the CALM and the axillary or inguinal freckling criteria, and both manifestations can be found in Legius syndrome. Facial dysmorphism is one of the variable clinical characteristics shared by all RASopathies.34 There were many patients in our sample with isolated features or a phenotype suggestive of RASopathy. But we only categorised patients that met the criteria for Noonan syndrome as NFNS cases. In 9 of the 12 patients genetic testing of the de NF1 gene was performed, confirming the diagnosis. Overall, these results suggest the presence of a characteristic phenotype in some children with NF1 that overlaps with other RASopathies, and as de Luca et al. stated, the NFNS type could be a variant of NF.35 The NFNS variant is most frequently caused by mutations in the NF1 gene. In fact, it has been proposed that testing for mutations in the NF1 gene is performed in patients that have both a Noonan phenotype and CALMs.35
In 83 patients, the disease was confirmed by means of genetic testing. In three patients, the results of direct testing for mutations were negative. These patients met two, three, and four criteria, respectively. We decided not to exclude them from the sample because they met the clinical criteria for NF1 and they did not exceed the percentage of false negatives reported for genetic testing.9,36 While other series have reported up to 40% of recurrent mutations in unrelated patients,37 we are describing 67 mutations (5 deletions and 62 mutations) in 67 different families. The mutations are distributed along the entire NF1 gene and most of them are frameshift and nonsense mutations, which were also the most frequent types found in other studies.37–39 Microdeletion syndrome has been associated with a particular phenotype10,40; in the 5 patients in our sample, the most salient features were general learning disability accompanied with varying degrees of intellectual disability and the presence of neurofibromas, but we did not find structural brain malformations, overgrowth, or malignancies.
ConclusionsThe data available on the phenotypical, radiological and genetic characteristics of 239 paediatric patients with NF1 that we present here provide an overview of the main findings and complications that characterise these patients in one of the largest cohorts to be found in the international literature. They evince that it continues to be difficult to make a clinical diagnosis at early ages. The proportion of patients diagnosed solely on the basis of cutaneous criteria, which cannot be distinguished from those of Legius syndrome, is even greater in children, so we suggest the possibility of combining both cutaneous diagnostic criteria (at least 6 CALMs with a diameter greater than 5mm in prepubertal children and greater than 15mm in pubertal children and/or presence of axillary or inguinal freckling) into a single criterion. While the need of performing diagnostic tests in asymptomatic patients remains subject to debate, cranial MRI can be very helpful in the clinical diagnosis of children with NF1 considering the high prevalence of ONG found in our series.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Duat Rodríguez A, Martos Moreno GÁ, Martín Santo-Domingo Y, Hernández Martín A, Espejo-Saavedra Roca JM, Ruiz-Falcó Rojas ML, et al. Características fenotípicas y genéticas en laneurofibromatosis tipo 1 en edad pediátrica. An Pediatr (Barc). 2015;83:173–182.
