


Introducción
La enfermedad de la orina con olor a jarabe de arce (MSUD) (OMIM 248600) es un trastorno en el metabolismo de los aminoácidos ramificados (BCAA) debido a la deficiencia del complejo multienzimático deshidrogenasa de los cetoácidos de cadena ramificada. Se caracteriza por un marcado aumento plasmático de la concentración de leucina, isoleucina y valina con presencia de aloisoleucina. Fue descrito inicialmente en 1954 por Menkes et al en cuatro pacientes que iniciaron un grave deterioro neurológico en la primera semana de vida y que fallecieron precozmente, y en los cuales su orina tenía un olor especial similar a los extractos de hojas de arce.
Es una enfermedad de herencia autosómica recesiva con una frecuencia media en todo el mundo de 1/185.000 recién nacidos, aunque existen grupos étnicos con mayor incidencia1,2. En Galicia, desde la introducción de la espectrometría de masas en tándem (MS/MS) en el Programa de Cribado Neonatal para alteraciones metabólicas, en junio de 2000, objetivamos una prevalencia de 1/39.300 (3/118.000).
Los principales fenotipos de MSUD incluyen la forma clásica (con una actividad enzimática descarboxilante inferior al 2 % respecto al control) que supone el 80 % de las formas de presentación, y la intermedia (actividad entre el 3 y el 30 %).
Hasta hace pocos años, los niños con jarabe de arce, sobre todo en la forma clásica, tenían muy mal pronóstico, pues la mortalidad era elevada y los que sobrevivían tenían graves repercusiones neurológicas.
Presentamos los tres casos de MSUD detectados tras la inclusión de la MS/MS en el Programa Gallego de Cribado Neonatal, explicando los progresos en el proceso diagnóstico y en el tratamiento en los momentos agudos de descompensación metabólica, claves para su desarrollo neurológico posterior.
Pacientes y métodos
Pacientes
Se estudian los tres niños (dos varones y una hembra) que fueron diagnosticados de MSUD en el período neonatal en Galicia durante los últimos seis años, entre julio de 2000 y julio de 2006. Los tres fueron controlados desde la detección hasta el momento actual en la Unidad de Trastornos Metabólicos del Hospital Clínico Universitario de Santiago.
En cada uno de ellos se evalúan la historia familiar y obstétrica, edad y síntomas al diagnóstico, concentraciones de aminoácidos ramificados y modo de tratamiento inicial (tratamiento conservador frente a medidas extracorpóreas). Para comprobar la eficacia de este tratamiento (velocidad de reducción de metabolitos tóxicos) se refleja en horas el tiempo que precisaron para que la concentración de leucina fuera inferior a 1.000 μmol/l.
Se realiza una valoración estricta del seguimiento evolutivo, reflejando la media y desviación típica de las concentraciones de leucina, valina e isoleucina anuales con el número de determinaciones por año. En función de las concentraciones de leucina se calcula la tolerancia dietética y se lleva a cabo valoración nutricional y del desarrollo psicomotor. Asimismo, se registraron las descompensaciones clínicas-analíticas que presentaron y que precisaron ingreso hospitalario, días totales de ingreso, concentraciones de aminoácidos ramificados (media y rango) durante esas descompensaciones y actitud terapéutica.
Se correlaciona el desarrollo psicomotor con el grado de afectación y concentraciones de leucina tanto al diagnóstico como en la evolución posterior.
En los tres se efectuó estudio enzimático y molecular.
Métodos
Cribado neonatal para MSUD por MS/MS en la muestra de sangre impregnada en papel (SS-903) tomada al tercer día de vida del recién nacido
El análisis se realiza sobre un disco de 3 mm de diámetro previa extracción con una solución de metanol que contiene, entre otros patrones internos, leucina y valina deuteradas; a continuación, se derivatiza con butanol-clorhídrico y se mide con un espectrómetro de masas (API 2000 Sciex). Aquellas muestras en las que los aminoácidos ramificados se encuentran por encima del punto de corte requieren confirmación diagnóstica mediante análisis de aminoácidos plasmáticos para comprobar la presencia de aloisoleucina3-6.
Análisis cuantitativo de aminoácidos
Se lleva a cabo por cromatografía de intercambio iónico (autoanalizador Biochrom 30) en la muestra de plasma, con desproteinización con ácido 5-sulfosalicílico, reacción poscolumna con ninhidrina y haciendo uso de patrón interno (norleucina).
El análisis de aminoácidos de cadena ramificada leucina, isoleucina y valina tambien se realiza a partir de la muestra de sangre impregnada en papel por cromatografía de intercambio iónico con condiciones cromatográficas e instrumentales similares a la muestra de plasma, pero con proceso previo de elución y desproteinización con ácido tricloracético al 3 %.
Ajuste dietético
Se llevó a cabo utilizando una base informática de confección propia que permite, tras la pesada de alimentos, obtener en 24 h el cálculo de calorías, proteínas naturales y totales, leucina, valina, lípidos y glúcidos referido a su peso.
Valoración antropométrica
Se determinó con los percentiles de peso, longitud y perímetro de cráneo.
Evaluación del desarrollo cognitivo y psicomotor
Se empleó la escala MSCA (escalas McCarthy de aptitudes y psicomotricidad) en los niños de edad preescolar y la escala de Brunet-Lézine en la niña de dos años. La puntuación del índice general cognitivo y/o el cociente de desarrollo están en el rango normal a partir de 85.
Estudio enzimático
Se midió la descarboxilación de leucina marcada ([1-14C] leucina) en fibroblastos cultivados frente a un control intraensayo.
Estudio molecular
Se realizó análisis de complementación genética y estudio de mutaciones en los genes BCKDHA y BCKDHB.
Resultados
Situación al diagnóstico
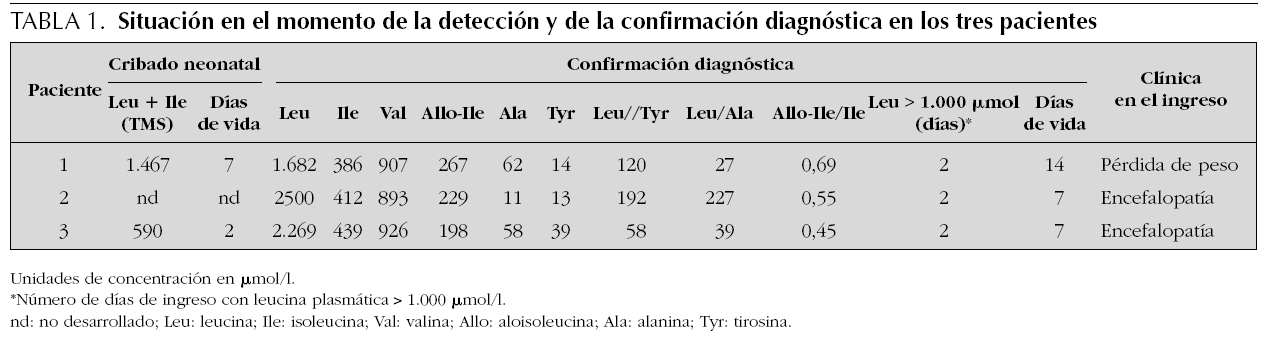
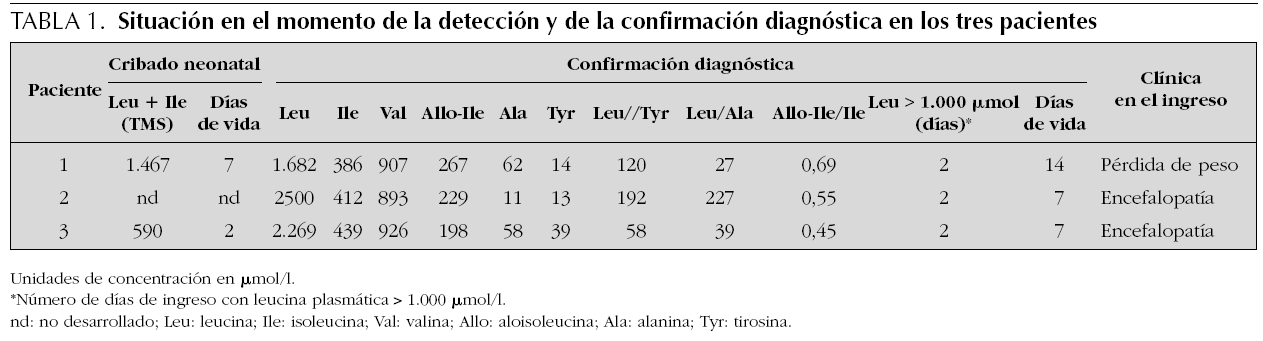
Como se refleja en la tabla 1, los tres niños fueron diagnosticados en el período neonatal; dos de ellos, en la primera semana de vida con clínica de encefalopatía aguda grave y concentraciones de leucina superiores a 2.000 μmol/l (2.500 y 2.269, respectivamente); el tercer paciente fue detectado por el Programa de Cribado Neonatal (leucina + isoleucina de 590 μmol/l en la muestra de dos días de vida) y el paciente 2 inicialmente sólo por la clínica. El paciente 1 fue detectado en el cribado metabólico por MS/MS con una concentración de leucina + isoleucina de 1.467 μmol/l en la muestra de sangre impregnada en papel tomada a los siete días de vida; en el momento de la confirmación diagnóstica, a los 14 días de edad, presentaba clínica de vómitos y succión lenta con pérdida de peso, pero no de distrés neurológico agudo tipo intoxicación; su concentración de leucina plasmática era de 1.682 μmol/l. Además, en la tabla 1 se muestran las concentraciones de los demás aminoácidos de interés en los tres casos.
Los tres neonatos fueron hijos de padres no consanguíneos, de etnia caucasiana, a término y con un embarazo y parto sin incidencias destacables.
Actitud inicial
En los dos niños con presentación neonatal grave, síntomas de encefalopatía-coma grave y concentraciones de leucina plasmática al ingreso superiores a 2.000 μmol/l, se indicó inicialmente fluidoterapia i.v. con alto aporte de glucosa y lípidos, manteniendo sodio entre 140-145 meq/l y suplemento de tiamina. Se llevó a cabo, además, depuración artificial mediante diálisis peritoneal, y en ambos casos se consiguieron a las 40 h valores en torno a 1.000 μmol/l7. Posteriormente se introdujo alimentación enteral exenta de aminoácidos de cadena ramificada (BCAA) (fórmulas alimentarias de Mead Johnson y SHS) con suplementos de isoleucina y valina (200-300 mg/día según concentraciones) y al quinto y cuarto días, respectivamente, alimentación con proteínas naturales hasta alcanzar progresivamente concentraciones de leucina inferiores a 400 μmol/l en ambos casos.
El tercer neonato (paciente 1), con leucina en plasma de 1.682 μmol/l a la confirmación diagnóstica, recibió desde el inicio fluidoterapia i.v. con glucosa y lípidos y alimentación enteral con leche exenta de aminoácidos de cadena ramificada. A las 48 h ya se introdujeron proteínas naturales con concentraciones de leucina de 290,7 μmol/l.
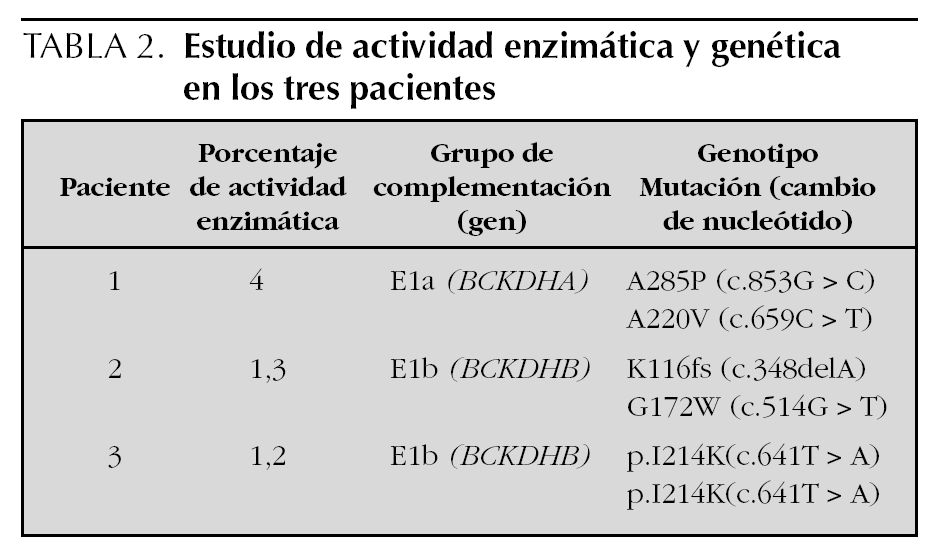
Estudio enzimático y genético (tabla 2)
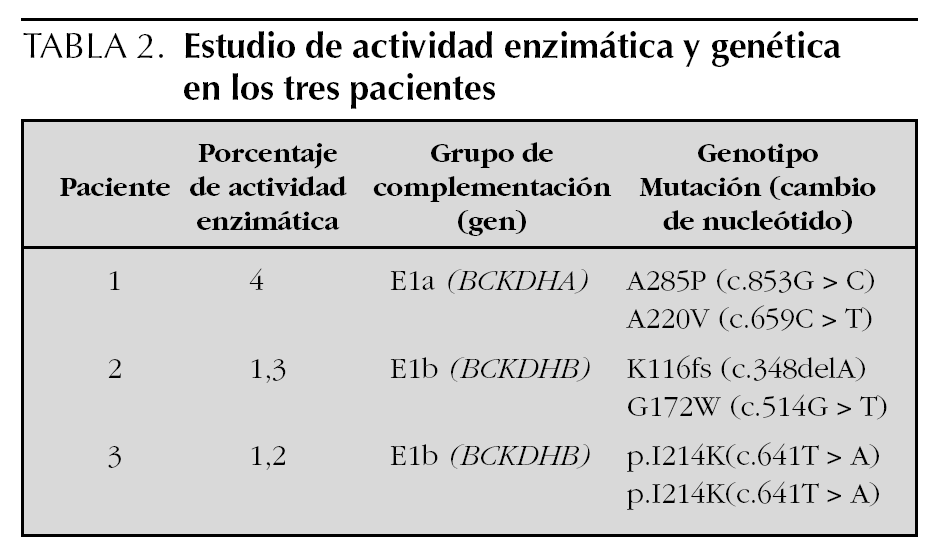
Se observó en el estudio enzimático una descarboxilación de leucina en fibroblastos muy deficiente (1,3 y 1,2 % respecto al control) en los pacientes 2 y 3, formas clásicas de MSUD. En el tercer niño, paciente 1, el cultivo de fibroblastos objetiva una descarboxilación de leucina del 4 % con respecto al control (forma intermedia de MSUD).
Los análisis de complementación genética indican que los niños con forma clásica pertenecen al grupo de complementación E1b (gen BCKDHB) y el niño con forma intermedia, al grupo E1a (gen BCKDHA). Estos estudios se efectuaron en el Centro de Diagnóstico de Enfermedades Moleculares de la Universidad Autónoma de Madrid.
Seguimiento
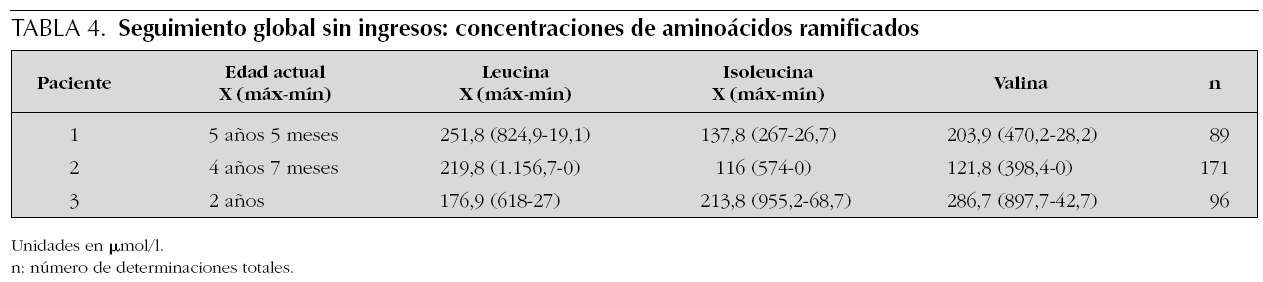
Como se puede observar en la tabla 3, las concentraciones medias anuales de leucina mantenidas tras el diagnóstico, excluyendo ingresos por descompensaciones, oscilan entre 156 y 356 μmol/l, con una frecuencia elevada de determinaciones al año. La situación global del seguimiento se muestra en la tabla 4, donde se observa la media de leucina con valores próximos a la normalidad, y el rango que sólo en un paciente sobrepasa los 1.000 μmol/l (1.156 μmoles/l).

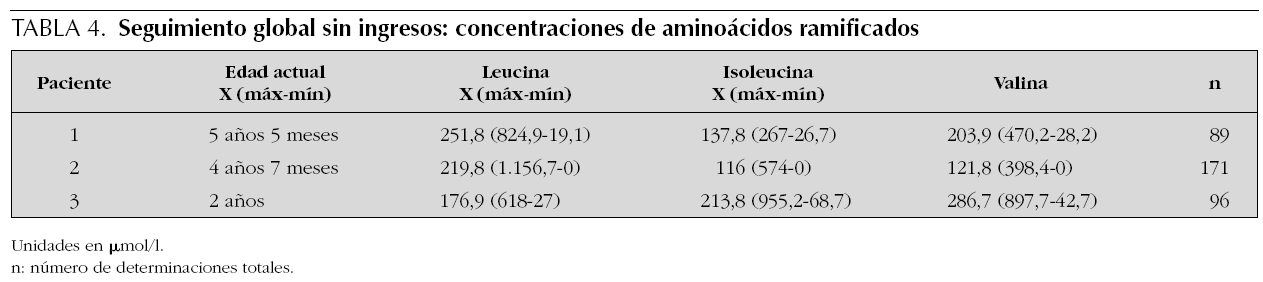
Los pacientes 2 y 3 con forma clásica neonatal mantienen evolutivamente concentraciones medias de leucina de 220 y 177 μmol/l; el paciente 1, de 251 μmol/l. La tolerancia actual de leucina es de 350 y 400 mg/día en los pacientes 2 y 3, y de 600 mg/día en el paciente 1.
Los tres presentaron alguna descompensación metabólica moderada que requirió ingreso hospitalario, siempre debidas a infecciones víricas y/o bacterianas (infección respiratoria y otitis fueron las más frecuentes). Durante estas descompensaciones, la concentración plasmática de leucina sólo superó esporádicamente los 1.000 μmol/l, el tratamiento siempre fue dietético, con suplementos de isoleucina, valina y tambien glutamina y alanina, y se pudo administrar en un episodio de gastroenteritis alimentación parenteral con una mezcla de aminoácidos exenta de leucina, valina e isoleucina (remitida desde la farmacia del hospital de Mänchen-Bogenhausen, Alemania). En la tabla 5 vemos reflejado el total de días de ingreso, el número de ingresos, el pico máximo de leucina alcanzado y el número de días totales con valores de leucina superiores a 1.000 μmol/l.

Respecto a su valoración antropométrica, los tres presentaron en todo momento percentiles en rango normal de peso, talla y perímetro de cráneo.
En cuanto a su evaluación neurológica, la puntuación del cociente de desarrollo fue elevada en el paciente con forma intermedia: 119; y de 87 y 94 en las formas clásicas.
Los dos niños de cuatro y cinco años acuden al colegio regularmente; la niña de dos años inicia este año la guardería.
Discusión
El paciente 1 fue detectado a partir del cribado neonatal por espectrometría de masas en tándem en la muestra de sangre impregnada en papel tomada al séptimo día de vida (las recomendaciones para la fecha de toma de muestra de cribado neonatal, en nuestra comunidad, estaban entre el quinto y el octavo día de vida en esas fechas)8, su confirmación diagnóstica e inicio del tratamiento tuvo lugar a los 14 días de vida, y no presentaba clínica de encefalopatía. La clínica precoz del paciente 2 sumada a las recomendaciones de la fecha de toma de muestra en ese momento hacen que su diagnóstico inicial sea realizado tras la clínica de encefalopatía-coma que presentaba. Desde el año 20028, la edad de la toma de muestra para cribado metabólico en Galicia se disminuye al tercer día de vida; esto permitió que la paciente 3 fuese detectada por medio del cribado neonatal en la muestra de sangre en papel con dos días de vida, por lo que ingresó a los siete días tras la confirmación diagnóstica de MSUD.
Dada la importancia de una actuación precoz, es de gran trascendencia la concienciación del personal sanitario implicado en el período neonatal, así como de las familias, para que la toma de la muestra se realice dentro de las recomendaciones. Es también muy importante el conseguir que la demora debida al envío de las muestras al laboratorio sea mínima.
Una actuación inmediata y correcta tras el diagnóstico es vital para un desarrollo neurológico posterior1,9,10. En nuestro caso, hemos recurrido a medidas dialíticas en los dos niños con clínica de encefalopatía-coma y hemos empleado la diálisis peritoneal por su fácil aplicación para lograr un descenso de la concentración de leucina por debajo de 1.000 μmol/l al segundo día de tratamiento. Se ha inducido anabolismo para eliminar el exceso de BCAA con nutrición parenteral y/o enteral con sonda nasogástrica continua y evitando y/o tratando la instauración del edema cerebral, sin técnicas de depuración extracorpórea con éxito en estos niños10-12.
Es importante, asimismo, durante la descompensación aguda mantener elevadas las concentraciones plasmáticas de valina e isoleucina, entre 400 y 600 μmol/l, (incluso algunos autores aconsejan entre 500 y 800 μmol/l)10 para aumentar el transporte de estos aminoácidos esenciales a los órganos, incluido el cerebro, ante la competencia con las concentraciones elevadas de leucina. Igualmente, se deben prevenir deficiencias de otros aminoácidos neutros que compiten con la leucina por el transporte de membrana dando suplemento de glutamina y alanina a 250 mg/kg/día. Las fórmulas y los aminoácidos i.v. usados en el tratamiento de MSUD tienen, en general, poco contenido de tirosina por su baja solubilidad, por lo que a veces hay que suplementarla manteniendo un cociente de leucina/tirosina menor de 5 (normal de 0,5 a 3,5) pues pueden presentar distonía aguda y coreoatetosis por su deficiencia2,10.
El seguimiento estricto a largo plazo, evitando o detectando precozmente las crisis de descompensación y manteniendo las concentraciones de leucina, como aminoácido guía del tratamiento de la enfermedad por ser el más neurotóxico, en valores lo más cercanos posible al rango normal (100-300 μmol/l) es también fundamental para conseguir un buen desarrollo cognitivo y psicomotor. Hasta hace poco se era más permisivo, aconsejando mantener concentraciones de leucina entre 200 y 700 μmol/l. En los últimos años, autores como Hoffmann et al13 recomiendan en los niños lactantes y preescolares que las concentraciones de leucina no excedan de 200 μmol/l para conseguir el mejor desarrollo intelectual posible.
Es muy importante una monitorización cómoda, fácil de realizar y que permita a los padres tomar la muestra directamente (determinación de BCAA por sangre impregnada en papel) para enviarla al laboratorio en el momento en que lo consideren oportuno con resultados rápidos con el fin de mantener los concentraciones de leucina en el rango aconsejado, consiguiendo, además, disminuir el número de visitas al hospital y mejorar su calidad de vida.
Aun así, las descompensaciones metabólicas son frecuentes en esta enfermedad, por lo que los padres, y más adelante el niño, deben estar entrenados para iniciar el tratamiento dietético mientras es trasladado y/o contacta con su centro de seguimiento. Es frecuente que tengan vómitos durante las descompensaciones, a pesar del tratamiento con ondansetrón, lo cual dificulta la tolerancia enteral, por lo que el disponer de una mezcla de aminoácidos i.v. exenta de aminoácidos de cadena ramificada aporta una mayor seguridad para el tratamiento de estos pacientes9,14.
Nuestros pacientes con jarabe de arce clásico, aunque tuvieron clínica de encefalopatía al diagnóstico, fueron tratados con prontitud y, evolutivamente, mantuvieron concentraciones de leucina por debajo del 300 μmol/l. Sólo uno de ellos (paciente 3) presentó una descompensación, con concentraciones de leucina superiores a 1.000 μmol/l (1.340 μmol/l). Sin embargo, sus índices generales cognitivos están en rangos normales aunque en límite inferior (87 y 94). Por el contrario, el paciente con forma clínica menos grave sin presentar encefalopatía al diagnóstico, evolutivamente mantuvo concentraciones medias algo más elevadas de leucina (240 μmol/l) y en las descompensaciones tuvo los picos mayores de leucina (hasta 1.579 μmol/l), pero su índice general cognitivo es de 119. Esto nos indica que tan importante como el seguimiento estricto es la situación al diagnóstico, sobre todo en ese período tan vulnerable de la vida como es el neonatal15.
Ninguno de nuestros pacientes respondió a la tiamina; sus mutaciones están en los genes E1a y E1b y no en el gen E2 que suelen tener los pacientes MSUD sensibles a la tiamina16,17. Hay más de 100 mutaciones descritas en el momento actual y aunque no se ha demostrado un significativo impacto de un locus mutante a otro en el fenotipo, las puntuaciones más bajas del índice general cognitivo parecen estar asociadas con mutaciones en el gen E1a16, que son las que presentan los pacientes 1 y 2 de nuestro estudio con la forma grave neonatal18-20.
Consideramos, a la vista de estas observaciones, que en los últimos años el pronóstico de la enfermedad ha mejorado notablemente, y ello va ligado a un diagnóstico precoz, a un tratamiento agresivo en la fase inicial y en las crisis de descompensación, así como el conseguir una mayor adherencia a la dieta sobre todo durante los primeros años de vida. Si el paciente sigue la dieta adecuadamente y hay un seguimiento clínico y bioquímico estricto, el trasplante hepático puede no ser la opción óptima en el tratamiento de la enfermedad de jarabe de arce10. El trasplante de células hepáticas y la terapia génica podrían ser una opción alternativa.
En conclusión, consideramos que incluir la enfermedad de jarabe de arce en los programas de cribado neonatal con una toma de muestra a los dos o tres días de vida permite mejorar notablemente el pronóstico de estos niños21. Los niños que fueron objeto de este estudio con concentraciones de leucina en valores cercanos al rango normal (de entre 100 y 200 a 300 μmol/l) en los primeros años de la vida mostraron una buena respuesta neurológica cognitiva.
La posibilidad de realizar determinación de BCAA en muestra de sangre impregnada en papel ha supuesto un importante avance en el tratamiento de los niños con jarabe de arce, ya que permite, ante la mínima posibilidad o sospecha de descompensación, que los padres puedan obtener una muestra y en pocas horas conocer las concentraciones de estos aminoácidos y de este modo acelerar la actitud terapéutica.
Disponer de una mezcla de aminoácidos intravenosos exenta de leucina, valina e isoleucina aporta una mayor seguridad en el tratamiento de estos pacientes.
Correspondencia: Dra. M.L. Couce Pico.
Servicio de Pediatría, 1C. Hospital Clínico Universitario de Santiago.
Choupana, s/n. 15706 Santiago de Compostela. España.
Correo electrónico: maria.luz.couce.pico@sergas.es
Recibido en diciembre de 2006.
Aceptado para su publicación en junio de 2007.
