


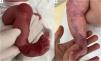
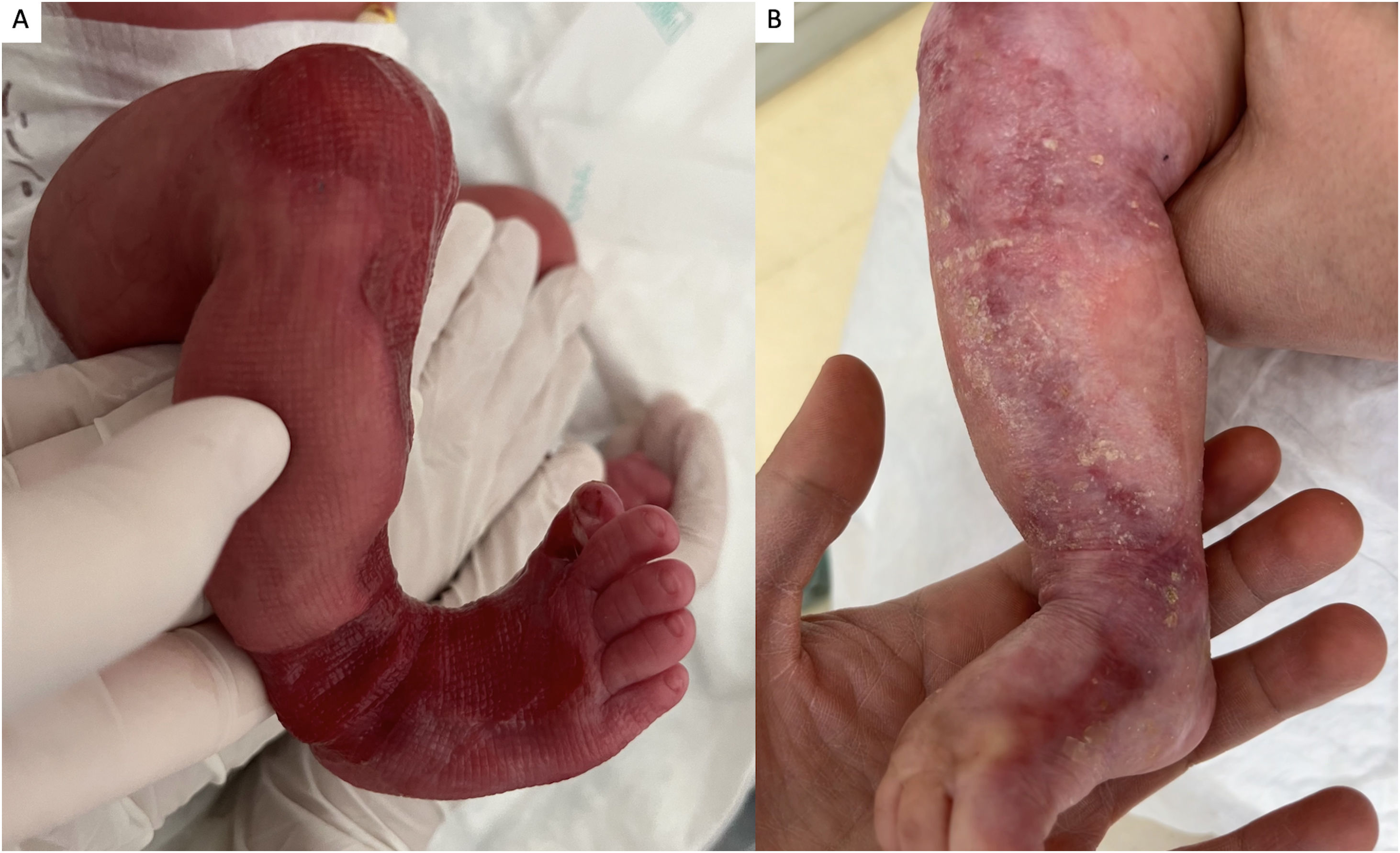
Un neonato de 10 días de vida fruto de padres no consanguíneos presentó ausencia congénita de piel en la extremidad inferior derecha al nacer. El examen físico reveló un área, en forma de S, de ausencia localizada de tejido cutáneo desde la rodilla a la cara anteromedial de la pierna, tobillo y pie derechos, así como distrofia ungueal en el primer dedo del pie y pequeñas erosiones en ambos pies (fig. 1). Sin afectación de la mucosa. Ausencia de alteraciones sistémicas. El padre y el abuelo paternos del paciente presentaban lesiones similares. El estudio genético detectó la presencia en heterocigosis de una variante patogénica de tipo missense (C.6191G>p-Gly2064Ala) en el gen COL7A1, alcanzándose el diagnóstico de epidermólisis ampollosa (EA) distrófica dominante con ausencia localizada de piel congénita (ALPC). Se recomendaron cuidados locales y vendaje de la herida, consiguiéndose la curación completa a los 5meses.
El síndrome de Bart1,2 es un trastorno infrecuente caracterizado por la asociación de aplasia cutis congénita localizada y que generalmente afecta a las extremidades inferiores, con EA y alteraciones ungueales2. También se ha descrito ALPC en localizaciones distintas de las extremidades inferiores, así como otras alteraciones extracutáneas2,3. La herencia de la EA asociada puede ser dominante o recesiva. Tradicionalmente, la EA distrófica ha sido la variante detectada con mayor frecuencia, aunque también puede haber asociación con la EA simplex o la epidermólisis ampollosa juncional2,3. La afectación ungueal no es necesaria para el diagnóstico. El síndrome de Bart se confirma mediante estudio genético, ocasionalmente apoyado por el mapeo antigénico y los hallazgos histopatológicos1,2.
