



Las anomalías endocrinológicas son frecuentes en los pacientes con deleción 22q11.2 e incluyen, por orden de frecuencia, hipocalcemia por hipoparatiroidismo primario, talla baja y disfunción tiroidea. Presentamos un caso de deleción 22q11.2 de diagnóstico tardío con afectación endocrina múltiple y diabetes mellitus tipo 1, y se revisan los conocimientos actuales de las manifestaciones endocrinológicas descritas en los pacientes con esta anomalía genética.
The endocrine abnormalities are common in patients with 22q11.2 deletion, and include hypocalcaemia due to primary hypoparathyroidism, short stature and thyroid dysfunction. We present a patient with delayed diagnosis of del22q11.2 who had multiple endocrine involvement and type 1 diabetes mellitus. A review is also made on the current knowledge of the endocrine manifestations described in patients with 22q11.2 deletion.
La deleción en diverso grado de la región 22q11.2 implica un espectro fenotípico tan amplio que determinadas entidades, actualmente englobadas dentro de esta anomalía (del22q11.2), habían sido descritas antaño como síndromes independientes, a saber, el de DiGeorge, el velocardiofacial, el conotruncal y el cardiofacial de Cayler.
El epónimo bajo el que se agruparon todas estas entidades, CATCH22, ha sido sustituido, con mejor suerte, por el de síndrome de deleción 22q11.21, si bien, parece mejor la denominación «espectro del22q11.2» teniendo en cuenta la amplia e impredecible variabilidad fenotípica que acabamos de referir.
Los problemas clínicos dependientes de esta pérdida de material génico en la región 22q11.2 tienen una prevalencia de un caso por cada 6000 nacidos vivos2. La cardiopatía constituye la anomalía más relevante y una de las más frecuentes (70-90%), habiendo coincidencia entre la mayor parte de las casuísticas en lo que respecta al tipo de defecto2, 3. Le siguen en frecuencia las anomalías velopalatinas (15-50%)2, 3, la hipocalcemia (21-72%), la agenesia tímica (hasta un 30%), los trastornos inmunológicos (10-55%) que pueden traducirse en un mayor riesgo para padecer infecciones y enfermedades autoinmunitarias2, 3, 4, y un fenotipo facial característico que se va acentuando con la edad y que puede incluir la típica facies asimétrica con el llanto por hipoplasia del músculo depresor del labio inferior3. Las anomalías neurológicas incluyen el retraso psicomotor y los problemas de aprendizaje (hasta un 80%), anomalías estructurales cerebrales (5-25%), diversos grados de espina bífida en menos del 10%2, 3, 5, crisis convulsivas2, 3 y trastornos psiquiátricos de diversa índole6. Otras anomalías menores son alteraciones nefrourológicas, esqueléticas, digestivas y oculares2, 3.
Las alteraciones endocrinológicas descritas, tema que nos ocupa, incluyen la hipocalcemia por hipoparatiroidismo primario, la talla baja y la disfunción tiroidea.
Pacientes y métodosAdolescente de 16 años de edad, con diabetes mellitus tipo 1, a la que durante una revisión rutinaria de hemoglobina glicosilada se le detecta una facies asimétrica con la sonrisa (Figura 1). Antecedentes personales: embarazo controlado. Parto por cesárea a la 38 semana con peso, longitud y perímetro en p10-25; se detectan mielomeningocele lumbosacro y ureterohidronefrosis izquierda de grado II con riñón izquierdo hipoplásico sin disfunción. Posteriormente se diagnostican: dificultades iniciales para la succión, debut de diabetes mellitus tipo 1 a los 4 años de edad (anticuerpos anti GAD65 e ICA positivos) y buen control metabólico posterior; dificultades de aprendizaje, estrabismo e hipotiroidismo subclínico no autoinmunitario (TSH 6-12μU/ml; VN: 0,4-5,5). Menarquia a los 12 años de edad. Talla < p3 (–2,3 DE) desde los 2 años de edad hasta 148,5cm (–2,6 DE) en la actualidad y considerada de adulto por una edad ósea de 18 años (Greulich & Pyle). Antecedentes familiares sin interés.
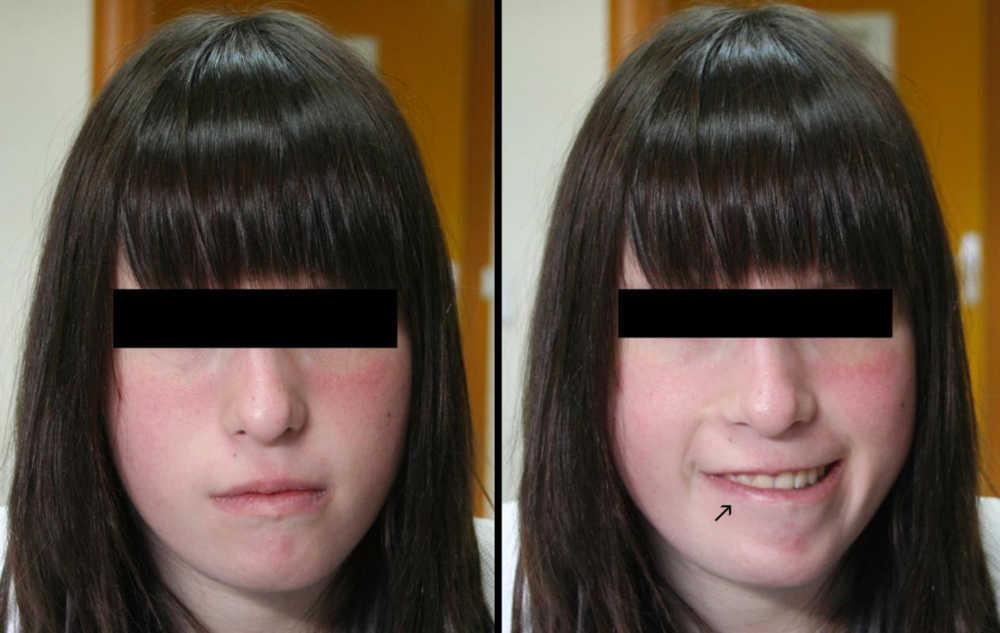
Figura 1. Facies en reposo y con la sonrisa.
Una exploración física más detallada permitió descubrir hallazgos fenotípicos peculiares como hendiduras palpebrales estrechas, raíz nasal prominente y cuadrada con punta bulbosa y narinas pequeñas (Figura 1). Este fenotipo facial y, más concretamente, la asimetría facial con el llanto o la sonrisa, asociados a las diversas malformaciones presentes desde el nacimiento permitieron la sospecha diagnóstica que se confirmó mediante MLPA (multiplex ligation-probe amplification), detectándose una pérdida de dosis en las sondas HIRA, CLDN5, KIAA1652, FLJ14360, PCQAP, SNAP29 y LZTR1, correspondientes a un tamaño de aproximadamente 3 Mb en la región 22q11. El cariotipo resultó normal así como el estudio genético realizado a los padres.
A partir de este momento, se procedió a la búsqueda de hallazgos clínico-analíticos y de imagen propios del espectro del22q11.2, encontrándose alteraciones cardiacas menores (arteria subclavia derecha anómala con pequeño divertículo de Kommerell; Figura 2) solo detectables mediante resonancia magnética cardiaca, insuficiencia velopalatina (problemas sutiles para la deglución con atragantamientos frecuentes) e hipoparatiroidismo primario. El diagnóstico de este último se basó en el hallazgo seriado de niveles bajos límites de PTH (10-16 pg/ml; VN: 12-65) y calcio (8,7-9,6mg/dl; VN: 8,8-10,6) junto con niveles de fósforo en el límite alto de la normalidad (4,4-4,6mg/dl; VN: 2,5-4,5).
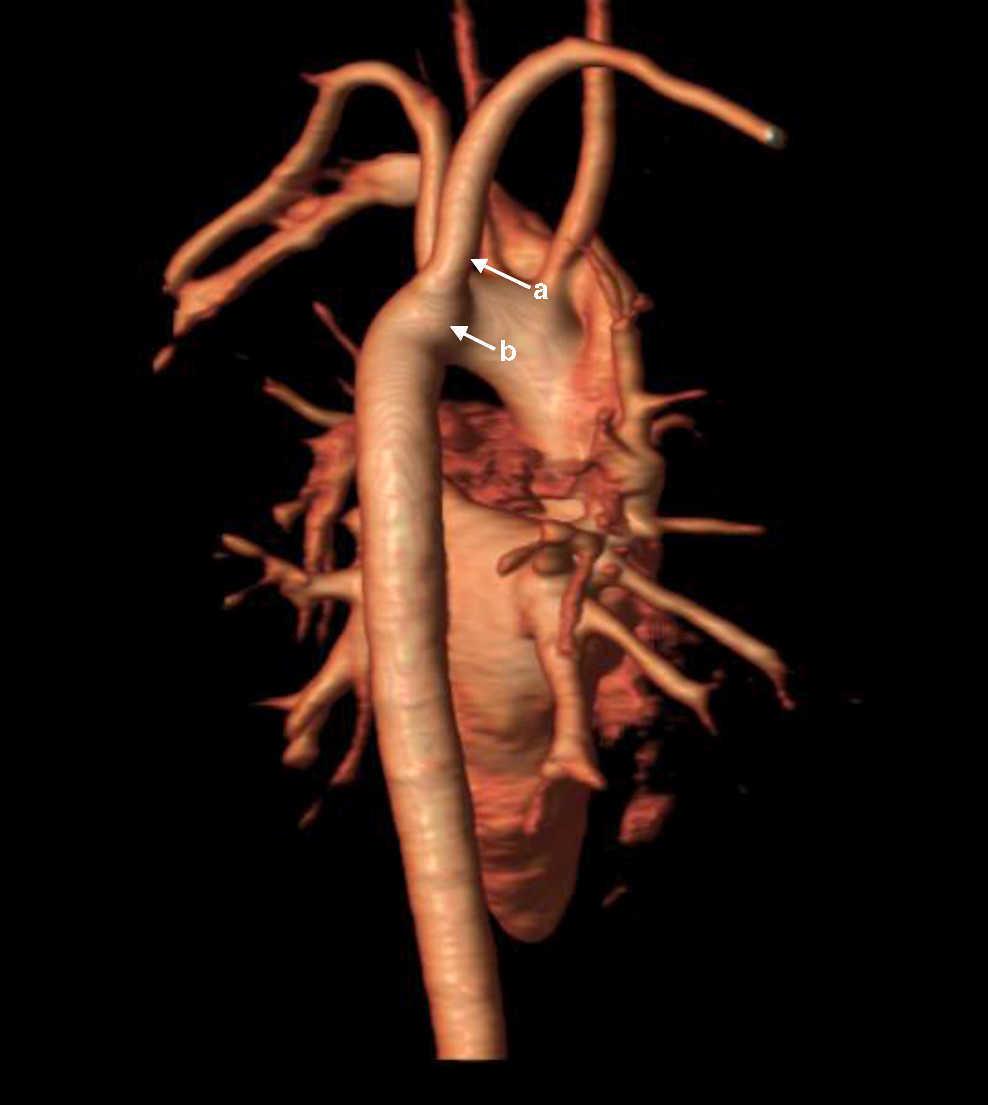
Figura 2. RM cardiaca 3D. Arteria subclavia derecha anómala (a) y divertículo de Kommerell (b).
El estudio de la inmunidad celular y humoral (subpoblaciones linfocitarias y respuesta proliferativa a mitógenos) resultó normal. Tampoco se encontró agenesia de timo.
DiscusiónHan sido numerosas las casuísticas publicadas que intentan describir detalladamente el espectro fenotípico que suponen las pérdidas de material génico de la región 22q11.2, llegándose a concluir en todas ellas la amplia variabilidad clínica existente y la consiguiente dificultad diagnóstica de los casos donde no está presente, o es mínima, la afección cardiológica2, 3, 7.
El caso presentado tiene esta particularidad al haberse retrasado el diagnóstico hasta los 16 años de edad y reunir varias de las manifestaciones endocrinológicas mencionadas en la literatura, algunas muy comunes y otras de reseña excepcional como la diabetes mellitus tipo 1.
El fenotipo facial y, más concretamente, la asimetría facial con la risa, resultaron ser los hallazgos que evocaron la sospecha diagnóstica, confirmada luego mediante MLPA. El estudio molecular negativo en los padres indica el origen de novo de la deleción, descrito así en el 95% de los casos presentados en casuísticas recientes3.
En lo que respecta a las endocrinopatías, se han descrito 4 cuya incidencia supera claramente a la de la población general, lo que justifica considerarlas consecuencia de la del22q11.2 (Figura 3). El presente caso merece especial atención por cuanto representa una rareza que combina los tres ejes hormonales típicamente descritos en esta deleción, a saber, el tiroideo, el paratiroideo y el crecimiento, añadiéndose la diabetes mellitus tipo 1 como entidad cuyo origen fisiopatológico dependiente de la anomalía génica se discutirá a continuación.
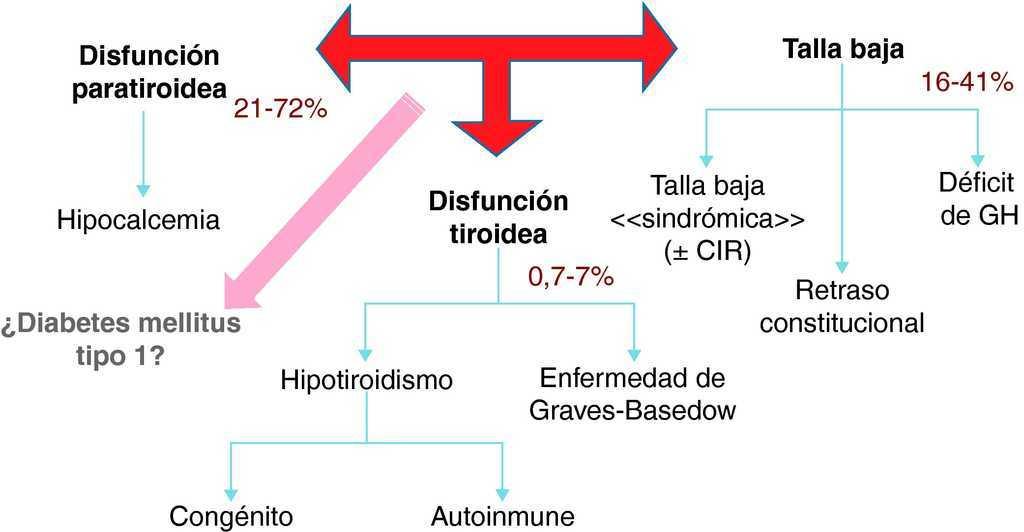
Figura 3. Anomalías endocrinológicas descritas en la deleción 22q11.2.
Hipocalcemia e hipoparatiroidismo primarioLa disfunción paratiroidea fue la primera endocrinopatía descrita en estos pacientes, siendo considerada como una de las manifestaciones cardinales del síndrome de DiGeorge.
Su prevalencia varía ampliamente según el criterio de selección utilizado (p. ej., el fenotipo)8; la Tabla 1 resume estas discrepancias (21%-72%).
Tabla 1. Prevalencias de las endocrinopatías asociadas a la deleción 22q11.2.
| Hipocalcemia/hipoparatiroidismo primario (21-72%) | |||
| Sin criterios de selección (del22q11 global) | Fenotipo como criterio de selección | ||
| Botto at al 2 | 21% | Fenotipo DiGeorge | 35-72% 8 |
| Ballesta et al 3 | 26%: (21% transitoria, 5% permanente) | ||
| Choi et al 13 | 32%: (24% transitoria, 8% permanente) | Fenotipo velocardiofacial | 13-22% 8,13 |
| McDonald-McGinn et al 11 | 49% | Fenotipo conotruncal | 10% 8 |
| Ryan et al 18 | 60%: (42% transitoria, 18% permanente) | ||
| Hipocrecimiento (16-41%) | |||
| Sin criterios de selección (del22q11 global) | Fenotipo como criterio de selección | ||
| McDonald-McGinn et al 11 | 41%: (36,9% talla baja «sindrómica», 4,1% déficit de GH) | Fenotipo velocardiofacial | 30% 12 |
| (20% RCC, 10% talla baja adulta) | |||
| Ryan et al 18 | 36% | ||
| Choi et al 13 | 16,4% | ||
| (13,1% CIR, 3,3% talla baja no CIR, 0% déficit de GH) | |||
| Disfunción tiroidea (0,7-7%) | |||
| Sin criterios de selección (del22q11 global) | Fenotipo como criterio de selección | ||
| Ryan et al 18 | 0,7% (solo hipotiroidismo) | Fenotipo DiGeorge | 5% 1 (solo hipotiroidismo) |
| Choi et al 13 | 3,3% | Fenotipo velocardiofacial | 0,8-7% 12,19 (solo hipotiroidismo) |
| (1,6% hipotiroidismo, 1,6% enfermedad de Graves) | |||
Este riesgo de hipocalcemia se debe, como se ha documentado en cirugías abiertas o autopsias de pacientes DiGeorge, a aplasia o hipoplasia paratiroidea9 y, de presentarse, su severidad clínica varía desde episodios de hipocalcemia neonatal grave y permanente, hasta formas transitorias, algunas de las cuales permanecen latentes y solo son evidenciables mediante pruebas de estímulo8.
Desde el punto de vista diagnóstico, se propone la búsqueda sistemática de hipoparatiroidismo primario en todos los casos8, 10. Por otra parte, Barisic et al7 en un estudio sobre grupos de riesgo de presentar del22q11.2 establecieron que un 33,3% de los pacientes pediátricos que habían sido investigados por hipocalcemia presentaban dicha anomalía genética, concluyendo la recomendación de hacer diagnóstico molecular a los pacientes que hayan presentado repetidamente hipocalcemia con PTH normal o baja sin un diagnóstico claro.
Talla bajaEl hipocrecimiento descrito en estos pacientes recoge tres posibles orígenes: talla baja «sindrómica» (TBS) con o sin retraso de crecimiento intrauterino (CIR), déficit de GH (DGH) o retraso constitucional (RCC).
La prevalencia obtenida entre diferentes casuísticas apenas difiere (16%-41%), aunque no queda bien definida la correspondiente a cada subtipo (Tabla 1). Por ejemplo, son escasas las casuísticas que recogen casos debidos a DGH (10% de tallas bajas según McDonald-McGinn), y donde un 50% presentaba anomalías en la imagen de resonancia magnética del área selar-supraselar (hipoplasia adenohipofisaria y ectopia neurohipofisaria)11. Las formas catalogadas de RCC, por recuperación de la talla final, representan un 10% de las tallas bajas de este síndrome12. Respecto a los CIR, solo Choi et al13 los recogen (26,2%), observando ausencia de catch-up en la mitad de éstos.
El caso presentado no requiere demasiado análisis pudiéndose catalogarse, según los datos aportados, de una TBS sin antecedente de CIR.
Disfunción tiroideaEs esperable que las anomalías tiroideas congénitas formen también parte de la del22q11.2 si se tiene en cuenta que el tiroides procede, en parte, de estructuras cuya embriogénesis depende de la integridad de la región 22q11.214. No obstante, se trata de la endocrinopatía asociada menos prevalente, con una tasa global que se sitúa en el 3,3%13, si bien, con diferencias sutiles entre casuísticas y según el criterio utilizado (Tabla 1). Lo mismo sucede con el tipo de disfunción, siendo más frecuente el hipotiroidismo (con o sin hipoplasia) que la enfermedad de Graves, aunque hay quienes los encuentran en igual proporción13. Y aunque casi todos los casos de hipotiroidismo primario, como el nuestro, son subclínicos o moderados y, por ende, descubiertos tardíamente, existen casos de presentación neonatal15. Por último, tampoco debe olvidarse que el riesgo inherente de autoinmunidad explica, al igual que la enfermedad de Graves, algunos casos de tiroiditis de Hashimoto13, 16.
Diabetes mellitus tipo 1Elder et al17 describieron en el año 2001 el primer caso de diabetes mellitus tipo 1 en un paciente con del22q11.2 atribuyéndose, sin poder demostrarse, a un defecto de la inmunidad celular que le conferiría mayor riesgo de autoinmunidad. Nuestra paciente representa el segundo caso publicado en la literatura, si bien no se han detectado alteraciones de la inmunidad que puedan explicar la conexión existente entre ésta y la del22q11.2. Dado que, como ya se ha expuesto, no se conoce bien la prevalencia de esta anomalía genética en la población general y teniendo en cuenta las grandes diferencias que existen para la de diabetes mellitus 1 pediátrica entre poblaciones, no se puede concluir si ésta representa una endocrinopatía más del espectro o se trata de mera coincidencia.
En resumen, el presente caso constituye una aportación poco habitual de endocrinopatía en los tres niveles descritos para la del22q11.2 junto con una diabetes mellitus tipo 1, hallazgos que sugieren con fuerza la necesidad de una evaluación endocrinológica completa en todos estos pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Recibido 24 Diciembre 2010
Aceptado 25 Enero 2011
Autor para correspondencia. jguerrerof@yahoo.es
