





Ivacaftor is a cystic fibrosis transmembrane conductance regulator (CFTR) potentiator that has been shown to improve the nutritional status and lung function of cystic fibrosis patients with the G551D mutation in clinical trials. The objective of this study was to describe the real-world progress of children receiving ivacaftor.
MethodsWe describe the real-world progress of four children with cystic fibrosis and the F508del/G551D genotype comparing data during ivacaftor treatment with baseline and with the year before commencing treatment.
ResultsOur sample comprised 4 children aged between 6 and 14 years and including one with a recent diagnosis of CF and other with persistent Mycobacterium abscessus (M. abscessus) and recurrent allergic bronchopulmonary aspergillosis. The baseline FEV1 was 58.5–81.8% of the predicted value, and ivacaftor was taken for a mean 24 months (range, 12–30 months). All patients experienced a significant and sustained improvement in lung function. Compared to baseline, the weight z-score improved by 1.53 points, and the BMI z-score by 1.6 points. Compared to the year before starting ivacaftor, the frequency of Pseudomonas aeruginosa (P. aeruginosa) isolates decreased (−0.4/patient/year), as did the number of respiratory exacerbations (−1.8/patient/year). The weight-adjusted dose of lipase per kilogram decreased progressively in all patients. In 1 patient, a previously persistent M. abscessus infection and recurrent allergic bronchopulmonary aspergillosis resolved during treatment.
ConclusionsChildren with cystic fibrosis and the F508del/G551D genotype receiving treatment with ivacaftor experienced a real-world improvement in lung function, nutritional status, respiratory exacerbations, isolation of P. aeruginosa, and dose of pancreatic enzymes.
Ivacaftor es un potenciador de la proteína reguladora de la conductancia transmembrana de la fibrosis quística (CFTR) que ha demostrado en ensayos clínicos mejoría del estado nutricional y la función pulmonar de pacientes con fibrosis quística con mutación G551D. El objetivo de este estudio es describir la evolución en la vida real de niños tratados con ivacaftor.
MétodosSe describe la evolución en vida real de 4 niños con fibrosis quística con genotipo F508del/G551D comparando los datos durante el tratamiento con ivacaftor respecto a la situación basal y al año previo al tratamiento.
ResultadosSe analizan 4 niños de entre 6 y 14 años, incluyendo uno con diagnóstico reciente de fibrosis quística y otro con infección persistente por Mycobacterium abscessus (M. abscessus) y aspergilosis broncopulmonar alérgica (ABPA) recurrente. El volumen espiratorio forzado en el primer segundo (FEV1) basal fue del 58,5–81,8% del predicho y recibieron ivacaftor 24 meses de media (rango 12–30 meses). Todos los pacientes tuvieron una mejoría significativa y mantenida de la función pulmonar. Respecto a la situación basal, el z-score del peso mejoró 1,53 puntos y el z-score del índice de masa corporal (IMC) 1,6 puntos. Comparado con el año previo al tratamiento con ivacaftor, disminuyeron la frecuencia de aislamientos de Pseudomonas aeruginosa (P. aeruginosa) (−0,4/paciente/año) y el número de exacerbaciones respiratorias (−1,8/paciente/año). La dosis de lipasa ajustada por kilo disminuyó progresivamente en todos los pacientes. Un paciente resolvió durante el tratamiento la infección por M. abscessus y la ABPA.
ConclusionesLos niños con fibrosis quística y mutación F508del/G551D tratados con ivacaftor mostraron en la vida real mejoría de la función pulmonar, el estado nutricional, las exacerbaciones respiratorias, los aislamientos de P. aeruginosa y la dosis de enzimas pancreáticas.
Cystic fibrosis (CF) is the most common life-limiting autosomal recessive disease in individuals of European descent. Its incidence generally ranges between 1 per 2000 and 1 per 6000 live newborns.1 The disease is caused by a mutation in the gene that encodes the cystic fibrosis transmembrane conductance regulator (CFTR) protein, which regulates the CFTR-dependent ion channel. More than 2000 CFTR mutations have been reported. The G551D mutation (Gly551Asp, c.1652G>A) affects up to 4% of patients worldwide.2,3 The CFTR channel is less likely to open in patients with this mutation, thus leading to considerably reduced chloride transport on the surface of epithelial cells.4
Ivacaftor is a CFTR potentiator that enhances chloride transport by increasing the likelihood of opening the CFTR channel in patients with class III mutations such as G551D.5,6 Two phase 3 studies have compared the usefulness of ivacaftor (at a dose of 150mg/12h) with placebo over 48 weeks in children aged less than 12 years and adults (STRIVE)7 and in children aged 6–11 years (ENVISION).8 In both studies, the ivacaftor-treated group experienced a significant improvement in FEV1, sweat chloride values and weight. A subsequent extension study (PERSIST) that included patients for a further 96 weeks of treatment confirmed the clinical improvement and did not report significant adverse effects.9
However, these studies present some of the typical limitations of clinical trials. Patients with conditions or microorganisms that could confound the study results were excluded, as were those with exacerbations or changes in treatment during the previous weeks and those who had received a cytochrome P450 3A inhibitor or inducer.
The objective of the present study was to analyze the progress of children who had received treatment with ivacaftor 2.5 years after the drug was first marketed in the United Kingdom.
MethodsThe study was performed at the Royal London Hospital, a designated CF center in East London that manages more than 100 children with CF. Followup at the clinic includes an evaluation of anthropometric values, pancreatic enzyme dosing, the number of courses of antibiotics, isolates of microorganisms, lung function, and clinical sample collection (sputum, cough swab). In addition, patients receiving ivacaftor underwent serial determination of serum transaminase levels and sweat chloride before starting therapy and at 6 weeks, 6 months, 1 year and 2 years of therapy. The predicted values for FEV1 (FEV1%), forced vital capacity (FVC%), and midflow (forced expiratory flow [FEF25–75%]) were based on Global Lung Function Initiative reference ranges. The z-score for weight, height, and body mass index (BMI) were based on data from the World Health Organization.
The study population comprised all patients aged less than 17 years with at least 1 G551D mutation who received ivacaftor between January 2013 and June 2015, irrespective of the duration of treatment. The dose administered was 150mg/12h daily or twice weekly if the patient was receiving a cytochrome P450 3A inhibitor.
We included all the data collected during treatment with ivacaftor, as well as data from the previous year when available. The analysis was based on the baseline value and the last value obtained during treatment with ivacaftor. In addition, we calculated the mean of the values recorded during treatment grouped into periods of 6 months. We compared these values with the baseline value (defined as the most recent value recorded before the first dose of ivacaftor) or with the first available value, and with the mean of the values from the year before treatment.
We performed the statistical analysis with the software SPSS version 19.0. We defined statistical significance as a p-value of less than 0.05. We used analysis of variance to compare the mean (and standard deviation [SD]) of the data grouped in periods of 6 months with that obtained during the previous year.
ResultsThe study population comprised 4 patients aged 6 to 14 years who had received ivacaftor for a mean of 24 months (12 months, 24 months, 30 months, 30 months). All 4 patients were heterozygous for F508del/G551D and had pancreatic insufficiency (Table 1). Two patients started ivacaftor when it was authorized in the United Kingdom in January 2013; patient 3 had to wait until he turned 6 in May 2013. The clinical situation of the patients at initiation of ivacaftor was very variable. Patient 1 had received a diagnosis of recurrent allergic bronchopulmonary aspergillosis (ABPA) yearly during the previous 5 years and experienced recurrence of ABPA in the first months of ivacaftor therapy, and ABPA was treated repeatedly with prednisone and itraconanazole or voriconazole as per protocol. In addition, 7 months before starting ivacaftor this patient was diagnosed with Mycobacterium abscessus lung disease having repeatedly grown Mycobacterium abscessus associated with a decline in lung function and computed tomography (CT) abnormalities. Eradication therapy had been prescribed with intensive IV, nebulized and oral antibiotic therapy in adherence with current guidelines. However, he remained Mycobacterium abscessus culture-positive prior to starting ivacaftor despite oral and nebulized continuation therapy. Due to concomitant treatment with clarithromycin and voriconazole during the first months, he was prescribed a reduced dose of ivacaftor to be taken twice weekly. On ivacaftor therapy, he became M. abscessus-negative and has not had any further episodes of ABPA. After 6 months, treatment with clarithromycin and voriconazole was discontinued and he commenced regular daily ivacaftor. He remains free of M. abscessus infection and ABPA at 21 months of ivacaftor therapy.
Clinical characteristics of the patients.
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | |
|---|---|---|---|---|
| Sex | Male | Male | Female | Female |
| Age at diagnosis | 2 years | 2 months | 2 years | 13 years |
| Sweat chloride at diagnosis, mmol/L | 126 | 96 | 99 | 90 |
| Genotype | F508del/G551D | F508del/G551D | F508del/G551D | F508del/G551D |
| Pancreatic insufficiency | Yes | Yes | Yes | Yes |
| Age at initiation of ivacaftor | 10 years | 7 years | 6 years | 14 years |
| Duration of therapy with ivacaftor | 30 months | 30 months | 24 months | 12 months |
| Microorganisms detected at initiation of ivacaftor | Mycobacterium abscessus | No | No | No |
| Number of Pseudomonas aeruginosa isolations in the year previous to ivacaftor therapy | 1 | 0 | 1 | No data |
Patient 3 did not have reliable spirometry results until 6 months after initiation of ivacaftor. Patient 4 presented at age 13 years and started ivacaftor once her genotype was confirmed shortly after diagnosis of CF, together with admission for intravenous antibiotics and commencing CF treatment. All 4 patients remain on ivacacaftor treatment at this time.
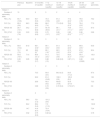
Lung function—especially FEV1 and midflow—improved in all 4 patients, and this improvement was maintained throughout the treatment period (Table 2). Baseline FEV1% ranged from 58.5% to 81.8%. When we compared the last lung function values recorded during treatment with the baseline values (or the first reliable value in the case of patient 3), we found an overall improvement in the percent predicted FEV1 of 25.72 percentage points. When we did the comparison with the mean from the year before initiation of ivacaftor in patients 1 and 2, the overall improvement in FEV1% was 18.85 percentage points. When we included all the lung function values obtained during treatment in the analysis, we found an overall improvement in the mean FEV1% of 11.96 percentage points with respect to baseline.
Lung function data before and during treatment with ivacaftor.
| Previous year | Baseline | 0–6 months of ivacaftor | 7–12 months of ivacaftor | 13–18 months of ivacaftor | 19–24 months of ivacaftor | 25–30 months of ivacaftor | Last spirometry | |
|---|---|---|---|---|---|---|---|---|
| Patient 1 | ||||||||
| Number of spirometry tests | 10 | 9 | 5 | 2 | 4 | 4 | ||
| FEV1 (%) | 64.7 (10.8) | 63.9 | 60.7 (5.6) | 70.4 (15.6) | 81.3 (4.0)* | 77.2 (2.0)* | 76.8 (1.5)* | 78.2 |
| FVC (%) | 85.9 (5.6) | 86.3 | 88.4 (6.6) | 86.2 (7.4) | 77.8 (5.8) | 78.0 (3.3)* | 76.4 (2.0)* | 77.5 |
| FEF25–50 (%) | 29.7 (14.1) | 34.2 | 27.1 (6.6) | 45.18 (30.3) | 86.1 (4.1)*** | 74.9 (11.1)*** | 77.0 (8.9)*** | 87.5 |
| FEV1/FVC | 0.64 (7.8) | 0.63 | 0.59 (6.8) | 0.70 (13.7) | 0.89 (2.2)*** | 0.85 (5.5)*** | 0.86 (1.7)*** | 0.86 |
| Patient 2 | ||||||||
| Number of spirometry tests | 10 | 2 | 3 | 3 | 2 | 3 | ||
| FEV1 (%) | 69.6 (10.2) | 81.8 | 88.1 (3.5)** | 89.1 (0.4)** | 81.4 (5.0)* | 87.3 (2.6)** | 89.8 (3.5)** | 93.8 |
| FVC (%) | 74.1 (7.5) | 85 | 89.2 (0)** | 89.1 (1.8)** | 82.5 (5.0)* | 87.0 (3.5)* | 87.8 (2.9)** | 90.4 |
| FEF25–50 (%) | 55.6 (19.7) | 71.4 | 80.6 (14.1)* | 91.3 (3.5)** | 72.5 (4.9)* | 84.4 (4.7)** | 95.4 (13.0)*** | 100.1 |
| FEV1/FVC | 0.83 (5.4) | 0.85 | 0.88 (3.7) | 0.88 (1.5) | 0.86 (0.8) | 0.87 (1.1) | 0.88 (2.926) | 0.90 |
| Patient 3 | ||||||||
| Number of spirometry tests | 1 | 2 | 3 | 3 | ||||
| FEV1 (%) | 70.3 | 64.6 (2.4) | 84.8 (4.3) | 84.5 (11.2) | 97.4 | |||
| FVC (%) | 92.2 | 81.2 (0.49) | 106.3 (5.6) | 105.2 (9.2) | 108 | |||
| FEF25–50 (%) | 26.6 | 40.0 (6.2) | 49.63 (15.6) | 50.9 (21.9) | 73.5 | |||
| FEV1/FVC | 0.69 | 0.72 (2.8) | 0.72 (5.6) | 0.72 (9.7) | 0.8 | |||
| Patient 4 | ||||||||
| Number of spirometry tests | 2 | 3 | ||||||
| FEV1 (%) | 58.5 | 75.1 (7.2) | 101.2 (5.9) | 108 | ||||
| FVC (%) | 84.2 | 89.4 (0.4) | 110 (11.3) | 122.9 | ||||
| FEF25–50 (%) | 14.7 | 41.4 (8.7) | 72.4 (15.4) | 72.4 | ||||
| FEV1/FVC | 0.62 | 0.75 (6.9) | 0.82 (4) | 0.78 | ||||
FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; FEF, expiratory flow.
Data are expressed as absolute values or as mean (SD).
Statistical significance was calculated with respect to the previous year:
The absolute improvement in the percent predicted midflow at the end of treatment was 46.65 percentage points with respect to baseline. The force vital capacity improved in 3 of the patients during treatment, whereas the FEV1/FVC ratio improved significantly in 1 patient. The absolute improvement in predicted FVC% at the end of treatment was 12.75 percentage points with respect to baseline.
Nutritional status, which we assessed by means of the z-score for BMI, improved steadily in all 4 patients during treatment with ivacaftor (Table 3). The absolute improvement in the z-score in the last anthropometric evaluation during treatment with respect to baseline was 1.6 points. When we grouped together the end-of-treatment values of patients 1–3 and compared them with those from the previous year, the mean z-score improved 0.50 points. When we included all the values obtained for patients 1–3 during treatment in the analysis, we found a similar improvement in the mean z-score (1.37 points with respect to baseline and 0.54 with respect to the previous year).
Nutritional data and doses of lipase before and during treatment with ivacaftor.
| Previous year | Baseline | 0–6 months of ivacaftor | 7–12 months of ivacaftor | 13–18 months of ivacaftor | 19–24 months of ivacaftor | 25–30 months of ivacaftor | Last | |
|---|---|---|---|---|---|---|---|---|
| Patient 1 | ||||||||
| Weight, z-score | 0.85 (2.44) | 0.50 | 0.59 (0.21) | 0.67 (0.46) | 1.27 (0.02) | 1.33 | 1.20 (0.11)* | 1.11 |
| BMI, z-score | 0.79 (0.07) | 0.68 | 0.46 (0.31) | 0.73 (0.64) | 1.5 (0.04)*** | 1.5 | 1.4 (0.14)** | 1.29 |
| Height, z-score | 0.61 (0.41) | 0.04 | 0.51 (0.05) | 0.30 (0.06) | 0.41 (0) | 0.52 | 0.43 (0.06) | 0.35 |
| Lipase/kg | 5416.7 | 6157.6 (304) | 5826.4 (273.1) | 4938.1 (118.4) | 3813.1 (502.3) | 3049.7 (231.3) | 3295.7 | |
| Patient 2 | ||||||||
| Weight, z-score | –0.23 (0.13) | –0.16 | –0.12 (0.07) | 0.2 (0.04)* | 0.11 (0.04) | 0.08 (0.23) | 0.32 (0.13)** | 0.41 |
| BMI, z-score | 0.34 (0.24) | 0.60 | 0.58 (0.10) | 0.9 (0.28) | 0.76 (0.28) | 0.65 (0.22) | 0.88 (0.18) | 1.01 |
| Height, z-score | –0.76 (0.16) | –0.98 | –0.91 (0.01) | –0.81 (0.05) | –0.80 (0.01) | –0.71 (0.12) | –0.62 (0.02) | –0.64 |
| Lipase/kg | 11,688.3 | 8462.3 (760.5) | 7577.9 (43.5) | 7187 (251.7) | 6849.7 (504.8) | 5371.4 (260.4) | 5187.3 | |
| Patient 3 | ||||||||
| Weight, z-score | –1.69 (0.32) | –2.15 | –1.22 (0.04) | –0.97 (0.01)* | –0.97 (0.04)* | –1.05 (0.06)* | –1.1 | |
| BMI, z-score | –0.12 (2.39) | –0.49 | 0.56 (0.01)* | 0.69 (0.01)** | 0.57 (0.25)* | 0.34 (0.12) | 0.21 | |
| Height, z-score | –2.16 (0.22) | –2.41 | –2.3 (0.03) | –2.18 (0.01) | –2.09 (0.20) | –2.01 (0.04) | –1.97 | |
| Lipase/kg | 13,312.7 | 13,003.2 (517.3) | 12,310.8 (5097.3) | 12,292.5 (1783.3) | 10,294.8 (225.9) | 10,135.1 | ||
| Patient 4 | ||||||||
| Weight, z-score | –3.31 | –0.72 (0.84) | 0.43 (0.15) | 0.6 | ||||
| BMI, z-score | –3.0 | 0.25 (0.91) | 1.38 (0.39) | 1.7 | ||||
| Height, z-score | –1.74 | –1.62 (0.09) | –1.20 (0.1) | –1.20 | ||||
| Lipase/kg | 4889.97 | 3618.31 (457.1) | 3252.03 | |||||
BMI, body mass index.
Data are expressed as absolute values or as mean (SD).
Statistical significance was calculated with respect to the previous year:
Our study also found an improvement in weight that remained constant throughout treatment. The improvement in the z-score for weight at the end of the treatment period was 1.53 points with respect to baseline and 0.50 points in patients 1–3 compared with the previous year. The improvement in the z-score for height was much more modest: 0.41 points at the end of treatment compared with baseline and 0.01 points in patients 1–3 with respect to the previous year.
The dose of lipase per kilogram of body weight fell significantly and gradually during treatment in all 4 patients (Table 3); when we grouped the values for patients 1 to 3, we found a 39.5% reduction in the dose at the end of treatment compared with baseline. In addition, the total dose of pancreatic enzymes decreased in 2 patients.
The sweat chloride value was 90mEq/L or greater in all 4 patients before starting ivacaftor. This value fell significantly during the first checkup at 6 weeks. In the 3 patients who had received ivacaftor for 2 or more years, the last determination revealed a value of less than 40mEq/L (Table 4). The mean reduction at the end of treatment was –58mEq/L.
The number of isolates of Pseudomonas aeruginosa in respiratory samples was generally low and fell slightly when compared with the year preceding initiation of ivacaftor in patients 1–3 (none of them had chronic infection). The mean number of isolates of P. aeruginosa was 0.67 per patient-year during the previous year (patients 1–3) and 0.27 per patient-year during the treatment period. The number of respiratory exacerbations requiring antibiotics also decreased in patients 1–3 with respect to the previous year: this was true for both oral agents (1.7 vs 3 courses per patient-year) and intravenous agents (0.5 vs 1 courses per patient-year).
Radiologic improvement was observed in 3 patients. The computed tomography images of patient 1 revealed a significant improvement, although the comparison was made at the end of treatment for M. abscessus infection and allergic bronchopulmonary aspergillosis. The radiograph of patient 2 was reported by the radiologist to show resolution of previous bronchial wall thickening in the left base when compared with baseline. Patient 4 had significant bronchiectasis at diagnosis, which improved significantly on treatment.
The only adverse effects observed were a mild and transient increase in transaminase levels in 1 patient (AST, 43IU/mL; ALT, 42IU/mL). There were no problems with adherence.
DiscussionIn this real-world study of 4 children with CF and the F508del/G551D genotype, ivacaftor led to an improvement in lung function, nutritional status, dose of pancreatic enzyme therapy, sweat chloride, number of isolates of P. aeruginosa and use of antibiotics. While previous studies on ivacaftor have also reported improvements in many of these parameters, real-world studies provide additional value with respect to effectiveness. On the one hand, clinical practice differs significantly from clinical trial protocols, and on the other, many patients are excluded from trials because they do not fulfill the inclusion criteria. In this sense, 3 of the 4 patients in the present study are not represented in clinical trials. Patient 1 received 9 months of therapy with cytochrome P450 3A inhibitors, which was an exclusion criterion in trials with ivacaftor.8 Patient 3, aged 6 years, did not have reliable spirometry values until 6 months after the start of treatment. Patient 4 had a late diagnosis and started ivacaftor together with a full multidisciplinary treatment regimen.
In the present study, we analyzed all lung function and anthropometric data recorded during treatment with ivacaftor and even included data from the previous year. In clinical trials, the protocol requires a baseline evaluation of lung function, the results of which are compared with those obtained after a previously established period, generally every 8 to 12 weeks. However, patients with CF frequently experience variations in lung function and body weight associated with respiratory exacerbations and various complications. In this sense, we believe that including all the data we collected during the treatment period adds value with respect to real-world effectiveness. Furthermore, drawing a comparison with values from the previous year can further improve the accuracy of the analysis, since the patient's situation at the start of treatment may not reflect the previous months.
The improvement in FEV1% with respect to the baseline value (25.72 percentage points) was higher than that observed in previous studies (10.6 and 12.5 points at week 24 and 10.5 and 10 points at 48 weeks8,9). Three of our patients had a baseline FEV1% of 70.3% or less, whereas most patients in the ENVISION and PERSIST studies had an FEV1% greater than 70%. In the ENVISION study, treatment was more effective in patients with more severe disease, expressed as a lower FEV1/FVC ratio (≤0.748) or baseline FEV1% (50–80%), with an improvement in FEV1% of around 20 points.8 In addition, the greatest improvement in FEV1% in our study was that observed in patient 4 (49.5 points), who started the multidisciplinary treatment program (including intravenous antibiotic therapy) when she started ivacaftor; therefore, in this case, some of this improvement could be related to the general CF care. However, the improvement seen in her CT scan is greater than would usually be expected. When we compared the last values of patients 1 and 2 with the mean for the year before starting ivacaftor, the absolute improvement in FEV1% was 18.85 percentage points, which is more in line with findings from clinical trials. The improvement brought about by ivacaftor may alter the natural decline in lung function experienced by patients with CF.10
The improvement in midflow was also greater than reported in clinical trials, probably for the same reasons as for FEV1, whereas the improvement in FVC was modest. The significant increase in the FEV1/FVC ratio in patient 1 was probably due to the improvement in bronchial obstruction after treatment of ABPA.
Nutritional status, as measured by BMI z-score, improved quickly and remained constant in all 4 patients during treatment. The improvement of 1.6 points was due partly to the improvement of 4.7 points in patient 4 that was in turn partly due to the multidisciplinary CF care and initiation of pancreatic enzyme replacement; when we excluded this patient from the analysis, the improvement was 0.57 points with respect to baseline and 0.50 points with respect to the previous year. These findings were similar to those of the ENVISION study at 48 weeks of treatment (compared with the placebo group).8 The changes in weight followed a pattern that was similar to that for improvement in BMI. However, the improvement in the z-score for height was very modest, and greater differences may have been observed if the treatment period had been longer or treatment had started earlier.
The weight-adjusted lipase dose decreased progressively in all patients during treatment, although the adjustment was based on clinical data without exhaustive monitoring of pancreatic function. The improvement could have been affected by the weight gain observed, although in 2 patients the total dose also decreased significantly. We might speculate that this effect was associated with the return to normal gastrointestinal pH values resulting from therapy with ivacaftor11 or with the improved patency of the pancreatic ducts. Of note, a significant improvement was observed in patient 4, despite the late initiation of treatment, possibly indicating that the changes observed in pancreatic function are not completely irreversible.
When data were compared with those of the previous year, P. aeruginosa was isolated less frequently during treatment, probably as a result of the improved mucociliary clearance produced by the treatment.11,12 In addition, the need for oral and intravenous antibiotics also improved. This is an important finding, since exacerbations have a significant impact on quality of life13 and are associated with a decrease in lung function that sometimes does not return to baseline values.14
Other studies have also reported the additional effects of ivacaftor on sinus disease, insulin secretion, hepatic steatosis and severe lung disease.15–20 Ivacaftor has also been shown to be effective against other class III mutations and in patients aged 18 or more years with the class IV mutation Arg117His-CFTR.21–23
We observed a radiological improvement in 3 patients during treatment. One patient cleared a M. abscessus infection and went onto remission from recurrent allergic bronchopulmonary aspergillosis, which may have contributed to the observed radiological improvement. Ivacaftor improves airway inflammation and mucociliary clearance, and radiological changes could be used as an objective marker for verifying individual response to treatment.24–26
The drug was well tolerated, and we did not detect any significant adverse effects from treatment. Postmarketing studies play a key role in ensuring the safety of the drug and monitoring potential long-term adverse effects in various clinical situations and in the presence of drugs with the potential for interactions. The safety of ivacaftor was recently verified in children aged 2–5 years, although increased transaminase levels were a common finding.27
Our study sample comprised only 4 patients, 1 of whom had received no more than 12 months of treatment at the time of writing this paper. Furthermore, it could be argued that the improvements in lung function and other variables were not solely related to ivacaftor treatment but also to initiation of multidisciplinary CF care (patient 4) and to resolution of M. abscessus and ABPA (patient 1). However, both patients continued to improve during the period of therapy. The objective of this study was not to address the exact figures of improvement, which are well described elsewhere, but to report the effectiveness and safety of ivacaftor in different and complex real-life clinical situations such as concomitant infections, drugs with possible interactions with ivacaftor and unstable patients. Moreover, we excluded the data for patient 4 from the comparative analysis with the year before the treatment.
The G551D mutation affects only 5% of patients in the United Kingdom. Since this limitation reduces the possibility of including a greater number of patients from a single CF unit, every effort should be made to perform real-world multicentre studies.
In conclusion, we performed a real-world study of 4 children with CF and the G551D mutation in whom treatment with ivacaftor led to a significant improvement in lung function, nutritional status, lipase dose, sweat chloride, isolation of Pseudomonas aeruginosa and use of antibiotics. Moreover, the effectiveness of the drug and its safety were verified in clinical situations that are not taken into account in clinical trials.
Conflicts of interestThe authors declare that they have no conflicts of interest
Please cite this article as: Gomez-Pastrana D, Nwokoro C, McLean M, Brown S, Christiansen N, Pao CS. Efectividad de ivacaftor en vida real en niños con fibrosis quística y mutación G551D. An Pediatr (Barc). 2019;90:148–156.
