



Hirschsprung Disease is caused by an impairment in cell migration from the neural crest to the gastrointestinal tract, resulting in an absence of neurons in the myenteric plexus. Many mutations in several genes have been associated to Hirschsprung disease; most of them affecting the RET proto-oncogen pathway. The purpose of this study is the description of novel and known mutations in genes associated to Hirschsprung disease and their prognostic implications.
Material and methodsRetrospective analysis of patients with Hirschsprung disease and positive genetic studies evaluated from 1970 to 2013.
ResultsWe found 21 positive genetic studies in the global series, 17 of them involving the RET proto-oncogene: Two of the mutations are novel and they have not been reported in the medical literature.
ConclusionsThe RET protooncogene is the main gene associated with Hirschsprung disease. There are still multiple unknown mutations related to the pathogenesis of the disease. The study of this gene must be part of the work-up of all patients with Hirschsprung disease, as well as their first degree relatives if the mutation is associated with MEN2A and MEN2B syndromes.
La enfermedad de Hirschsprung está causada por un defecto de migración celular desde la cresta neural hasta el tracto gastrointestinal, resultando en la ausencia de neuronas en el plexo mientérico. Mutaciones en varios genes han sido asociadas a la enfermedad de Hirschsprung, la mayoría afectando a la vía del protooncogén RET. El objetivo de este estudio es la descripción de mutaciones tanto descritas como nuevas asociadas a la enfermedad de Hirschsprung, así como sus implicaciones pronósticas.
Material y métodosAnálisis retrospectivo de pacientes con enfermedad de Hirschsprung y resultados genéticos positivos desde 1970 hasta 2013.
ResultadosEn la serie global, 21 pacientes tenían resultados genéticos positivos, 17 de ellos afectando a la vía del protooncogén RET: 2 de las mutaciones son nuevas y no han sido previamente descritas en la literatura médica.
ConclusionesEl protooncogén RET es el principal gen asociado a la enfermedad de Hirschsprung. Todavía hay múltiples mutaciones desconocidas relacionadas con la patogenia de la enfermedad. El estudio genético del gen RET debe formar parte del estudio diagnóstico de todos los pacientes con enfermedad de Hirschsprung, así como de sus familiares de primer grado en caso de que las mutaciones estén asociadas a los síndromes MEN2A y MEN2B.
Hirschsprung Disease (HSCR) is caused by an impairment in cell migration from the neural crest to the gastrointestinal tract, resulting in an absence of neurons in the myenteric plexus.1,2 The global incidence of HSCR is about 1 in 5000 births,3 and it is most frequent in Asians, followed by African-Americans and Europeans. In approximately 80% of patients, the disease presents in the short-segment (or rectosigmoid) form. In 15% of patients, the absence of neurons extends proximally, resulting in the long-segment form of the disease. In the remaining 5% of cases, there is involvement of the whole extension of the colon.4 Furthermore, there are infrequent cases that have further involved into the thin bowel. Patients usually debut with impairment of the intestinal motility, presenting with obstruction, distension, constipation or failure to pass meconium. The diagnosis is confirmed by histological analysis, based on the presence or absence of ganglion cells in the myenteric plexus. Treatment is surgical and consists in the resection of the affected part of the bowel, with posterior pull through of the ganglionic portion of the colon and end-to-end anastomosis.5,6
Several gene defects are associated with HSCR.7 Genetic causes are identified more frequently in the long-segment forms of the disease compared to the short-segment forms.8,9 Mutations in the RET proto-oncogene have been identified in several families with HSCR, a region currently known as Hirschsprung disease 1 susceptibility locus HSCR1, OMIM 142623. Other loci for isolated HSCR have been mapped. For instance, HSCR2 OMIM 600155 is associated with changes in the EDNRB gene on 13q22; HSCR3 OMIM 613711 is associated with changes in the GDNF gene on 5p13; HSCR4 OMIM 613712 is associated with changes in the EDN3 gene on 20q13; HSCR5 OMIM 600156 has been mapped to 9q31, HSCR6 OMIM 606874 to 3p21, HSCR7 OMIM 606875 to 19q12, HSCR8 OMIM 608462 to 16q23 and HSCR9 OMIM 611644 to 4q31-q32. On the other hand, in some patients HSCR manifests in the context of a syndrome,10 such as Bardet-Biedl syndrome, Down syndrome,11 Fryns syndrome, Goldberg-Shprintzen syndrome, congenital central hypoventilation syndrome,12 intestinal neuronal dysplasia type B, Mowat-Wilson syndrome,13 MEN2A, MEN2B,14 neurofibromatosis type 1, Pitt-Hopkins syndrome, Smith-Lemli-Opitz syndrome or Waardenburg syndrome type 4. In addition, approximately 18% of patients with HSCR also have associated congenital anomalies.15 These are most frequently cardiac abnormalities (such as atrial septal defect, ventricular septal defect, patent ductus arteriosus, tetralogy of Fallot), gastrointestinal abnormalities16 (intestinal malrotation, imperforate anus, Meckel’s diverticulum, anorectal fistula), genitourinary abnormalities17 (cryptorchidism, inguinal hernia, hypospadias, renal malformation, urethral fistula) and central nervous system abnormalities (intellectual disability, Dandy-Walker malformation, microcephaly).
The goal of our study was to describe the mutations found in patients with HSCR managed in a tertiary care hospital that had undergone a complete genetic evaluation, determine whether they were novel or known, and discuss the clinical relevance and prognostic implications of genetic testing in the management of HSCR.18
MethodsWe made a retrospective review of the health records of all paediatric patients with HSCR diagnosed between 1970 and 2013. We included patients with histological confirmation of the disease; this is, with evidence of absence of ganglion cells in the myenteric plexus on the bowel biopsy. We considered hypertrophy of cholinergic nerve fibres, increased acetylcholinesterase activity in the muscularis mucosae or decreased calretinin immunoreactivity in the lamina propria histological findings supportive of the diagnosis but not pathognomonic.
The study adhered to the ethical principles of the Declaration of Helsinki (Tokyo revision, October 2004) and with regulations concerning patient confidentiality. We obtained the approval of the Ethics Committee of our hospital before starting the study, as well as the informed consent of the parents of the 3 patients included in the analysis.
The study focused on those patients whose health records featured positive results of genetic testing. We retrieved clinical and demographic data and compared them to the results of genetic testing, with particular emphasis on novel mutations.
We compared the mutations found in the patients with those described in the medical literature and in the following genetics databases: DMDM (Domain Mapping of Disease Mutations), BioMuta 2.0, UniProt, COSMIC, ClinVar (Clinical Variants), LOVD (Leiden Open source Variation Database), EXAC (Exome Aggregation Consortium).
The genetic testing methods used varied through the years under analysis. From 1970 to 2007, karyotyping was the only test performed. Between 2007 and 2011, array-based comparative genomic hybridization (aCGH) and RET analysis were introduced. Since 2011, other genes (when suspected) have been analysed directly by means of Sanger sequencing or using gene panels for Mendelian diseases (clinical exome sequencing).
Genetic analyses of DNA were performed in samples of peripheral blood lymphocytes by amplification and subsequent sequencing by polymerase-chain reaction (PCR) of the coding regions and exon-intron junctions of the RET gene. The screening of the coding sequences and intron/exon boundaries of RET (NM_020975.4) was performed by direct sequencing. Primers were designed with the help of Primer 3 v0.4.0 software (http://bioinfo.ut.ee/primer3-0.4.0/) and SNPCheck V3 (https://secure.ngrl.org.uk/SNPCheck/snpcheck.htm). The PCR products were sequenced using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems; Foster City, CA, USA) and run on an ABI3730XL Sequencer (Applied Biosystems, Foster City, CA). Conservation, in silico pathogenicity prediction and control population frequency analysis (Exome Aggregation Consortium [ExAC]) of the identified RET variants was carried out using Alamut V2.6–1 software (Interactive Biosoftware, Rouen, France) and MutPred (http://mutpred.mutdb.org/). We also consulted the RET mutation database19 to check whether any of the detected variants had been described previously, which would support its pathogenicity.
The study of genomic rearrangements involved karyotyping and/or the use of aCGH. The PHOX2B gene was analysed either with either direct Sanger sequencing or clinical exome sequencing. We applied the recommendations and guidelines of the American College of Medical Genetics and Genomics.20
ResultsIn the period under study (1970–2013), there was clinical suspicion of HSCR in 193 patients. The diagnosis of HSCR was confirmed histologically in 160 patients. The male-to-female ratio was 113:47. The age at diagnosis ranged from 0 months to 7 years.
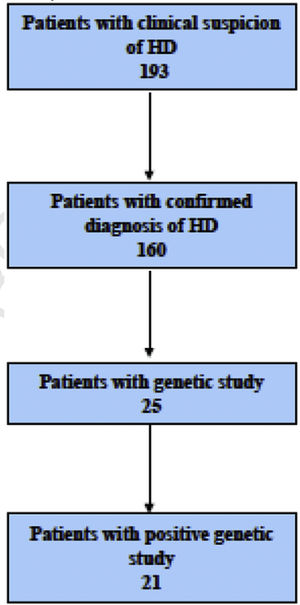
We did not find documentation of genetic testing for most patients, probably because it was not part of the routine workup for HSCR until recent decades. We found the first patient that underwent genetic testing in 1989. Twenty-five patients had undergone genetic testing, which detected changes in 21 patients.
In 17 cases (80.95%), the changes were mutations in the RET proto-oncogene; one was a benign variant in the RET proto-oncogene (2608-24 G>A; 4.76%)21 and one a mutation in the PHOX2B gene22,23 (4.76%). Two patients had genomic disorders: one had trisomy 21 (4.76%), and another trisomy 22 (4.76%) (Fig. 1).

Of the 17 patients with mutations in the RET proto-oncogene, 13 had mutations previously reported in the literature. Two patients had a combination of 2 mutations (c.1083C>T;p.R360W and c.215T>C;p.L72P). Thus, a total of 11 different mutations were detected in our patients (Table 1).
Genetic findings in RET gene.
| Mutation sequence | Description |
|---|---|
| c.988C>T; p.R330N | Association with HSCR described in the literature |
| c.107A>G;p.Y36C | Association with HSCR described in the literature |
| c.1083C>T;p.R360W | Association with HSCR described in the literature |
| c.428 C>G; pA143G | Association with HSCR described in the literature |
| 987dupT; pF329FfsX24 | Association with HSCR described in the literature |
| c.1252C>T;p.R418X | Association with HSCR described in the literature |
| c.1859G>A;p.C620Y | Association with HSCR described in the literature |
| G>A;p.D300N | Association with HSCR described in the literature |
| c.1831T>G;p.C611G | Association with HSCR described in the literature |
| c.215T>C;p.L72P | Not previously described in the literature |
| c.367C>T; p.L123F | Not previously described in the literature |
Nine of the 11 mutations (81.82%) had been registered previously in databases as either being pathogenic or increasing the risk of developing the disease.
Two (18.18%) of the 11 mutations (c.367 C>T; p.L123F and c.215T>C; p.L72P) have not been previously described in the literature and are not included in any of the databases.
Clinical findings in the patients with novel mutations and the patient with a PHOX2B mutationPatient 1 (c.367C>T; p.L123F)Boy born preterm product of a dichorionic diamniotic twin pregnancy. Apart from the gastrointestinal (GI) symptoms, the patient had multiple small-sized atrial septal defects (ASDs) that closed spontaneously by age 4 years. He did not have any other relevant medical history.
At 24h of life, the patient exhibited abdominal distention, vomiting and constipation. At 5 days post birth, he received a diagnosis of caecum perforation, which was treated with a right hemicolectomy with terminal ileostomy followed by reconstruction with ileocolic anastomosis at the age of 7 months. After surgery, the patient experienced multiple episodes of bowel obstruction, which were initially managed medically, until biopsy samples were performed to evaluate the recurrent obstruction. The findings in the biopsy samples were compatible with HSCR in the rectum, sigmoid colon, descending colon and splenic flexure (absence of ganglion cells in the myenteric plexus, although without hyperplasia of cholinergic neurons). This confirmed the diagnosis of long-segment HSCR. These findings led to performance of a colostomy. Genetic testing revealed a mutation in the RET gene (c.367C>T; p.L123F). Testing was thereon extended to his first-degree relatives. The same mutation was found in the patient’s father and grandfather, both of who were asymptomatic.
One year later, the patient presented with constipation through the colostomy. New biopsy samples were collected, and a new colostomy was placed 14cm proximal to the previous one. At age 3 years, the patient was referred to our center to undergo an elective Swenson pull-through procedure with a protective ileostomy, which was closed 6 months later. During the outpatient follow-up, mildstenosis of the anastomosis was detected, confirmed by colonoscopy and successfully treated with dilatations.
Despite the above, the patient suffered a severe episode of enterocolitis at the age of 4. It was managed conservatively, and the discharge summary recommended daily rectal stimulation and irrigation.
At the time of this writing, the patient is 7 years old, attends school, and has achieved the milestones expected for his age. His physical development is adequate. He has total control of sphincters and has daily bowel movements, although he receives an enema once a week with the purpose of preventing further episodes of enterocolitis.
Patient 2 (c.215T>C; p.L72)Girl born at term (37 weeks) with adequate weight for gestational age (2950g) in an uncomplicated delivery. Apart from HSCR, she had a past history of patent oval foramen. The patient presented at 48h of life with signs and symptoms of bowel obstruction (abdominal distension, bilious vomiting, and delayed passage of meconium). The obstruction resolved after passage of meconium 36h after. During the following months, the patient had anorexia and malnutrition, in spite of normal bowel movements. At 5 months of age she presented with fever, abdominal distention and food refusal. An abdominal X-ray revealed pneumoperitoneum and dilation of intestinal loops. The patient required surgery and underwent an ileostomy; a caecal perforation was detected and closed during the procedure. In addition, biopsy samples were obtained from the rectum, sigmoid, descending, transverse and ascending colon, as well as the cecum and ileum, all of which had characteristics compatible with HSCR, thus confirming the diagnosis of long-segment disease. Biopsy samples were also taken from the rest of the small bowel, but those had normal histology. Genetic testing revealed that a mutation in the RET gene (c.215T>C; p.L72).
At 22 months of age, the patient underwent an elective abdominoperineal pull-through performed with the Swenson technique with placement of a new ileostomy, which was closed 5 months later. In the months that followed the surgery, the patient had occasional vomiting, poor oral intake due to oral intolerance and poor weight gain, and required nocturnal continuous enteral nutrition in subsequent months. At the time of the latest check-up, she was 14 years old and exhibited age-appropriate development. Due to her difficulty to tolerate oral nutrition, she has remained in follow-up by the nutritional rehabilitation team.
The patient’s younger sister also had the same RET mutation, which was inherited from their mother. Their father’s genetic evaluation did not detect any abnormalities. The sister was born at term, with passage of meconium at 8h of life. She has no relevant past history. She presented at 28h of life with bilious vomiting and food refusal. She underwent an exploratory laparotomy with collection of biopsy samples, leading to detection of long-segment HSCR, in this case limited to the descending colon. This patient was treated with an elective abdominoperineal pull-through with the same technique at age 15 months. The ileostomy was closed 4 months later. After the surgery, she developed a supraumbilical eventration over the laparotomy scar in the midline, which did not cause complications and was surgically repaired at 3 years of age.
After the surgery, the main problem in this patient was low weight gain, as had occurred with her sister. She also had 2 episodes of enterocolitis, presenting with fever, vomiting, abdominal pain and constipation. Both were medically managed.
At present, she is 8 years old, with good bowel management, and under follow-up in the outpatient clinic.
Patient with a PHOX2B mutation (c.367C>T; p.L123F)The patient with a PHOX2B mutation was boy delivered at term with adequate gestational weight following an uncomplicated pregnancy. Aside from HSCR, he had a relevant history of right nephrolithiasis with secondary hydronephrosis, congenital central hypoventilation syndrome (nocturnal hypoventilation) and cow’s milk protein allergy.
At 12 days of life, the patient presented with abdominal distension, nausea and vomiting. He received a diagnosis of complete intestinal obstruction requiring urgent laparotomy with colostomy at the level of the hepatic flexure. There were no immediate post-operatory complications, and therefore the patient was discharged with the colostomy. During the neonatal period, congenital central hypoventilation syndrome was suspected, leading to genetic testing. The latter detected a heterozygous mutation in the PHOX2B gene. The patient was given a diagnosis of congenital central hypoventilation syndrome and managed with placement of a tracheostomy tube and home mechanical ventilation.
At 5 months of life, the patient was readmitted for another episode of intestinal obstruction, with findings of imaging tests indicative of gastric volvulus (later determined to be caused by surgical adhesions). The patient underwent a gastropexy and, since Hirschsprung disease was suspected at this point, colon biopsy samples were collected during this procedure. The histological examination revealed absence of ganglia in the myenteric plexus and mild hyperplasia of cholinergic neurons in the samples obtained from the rectum and the sigmoid colon, leading to diagnosis of short-segment HSCR. At this time, the patient was scheduled for an elective pull-through procedure. The first intervention took place when the patient was 11 months old, but a second intervention was required, since biopsies from the remaining colon were found to be aganglionic after the first procedure. Shortly after the second procedure, the colostomy was removed.
In subsequent months, the patient required multiple rectal dilatations due to anastomotic stricture, and presented severe perianal and perineal dermatitis, requiring a new ileostomy. The disease course in this patient was also complicated by chronic malnutrition (most likely a combination of HSCR, cow´s milk protein allergy, and congenital central hypoventilation syndrome). The patient was referred to our center at 7 years of age for a third abdominoperineal descent. After this last descent the patient had a favorable outcome and the ileostomy was removed 3 months later.
Patient is currently aged 9 years. He requires daily rectal irrigations and ocassionally defecates spontaneously. He temporarily required nocturnal enteral nutrition, but currently is able to do most of his intake orally. He was diagnosed with cognitive deficiencies for his age and a delay in speech, which has greatly improved since not requiring so many hospitalizations.
DiscussionThis historical series from 1970 to 2013 shows the progress in the knowledge of the genetic mutations associated with HSCR and their clinical repercussions, which has allowed us to improve prenatal counselling, as well as diagnostic and prophylactic measures in relatives of affected individuals.
The rearranged during transfection gene (RET), located at 10q11, is the gene most strongly associated with HSCR, as described in the medical literature and evidenced in our study. It is a tyrosine kinase receptor that regulates the action of growth factors on enteric neurons. It is therefore the first gene that should be included in the genetic evaluation when HSCR is diagnosed. In our series of patients, it was the most prevalent finding of genetic testing.
Paired-like homeobox 2b (PHOX2B) is a gene located in chromosome 4 that encodes a transcription factor expressed in neural crest cells that can interfere with expression of the RET gene. In this series, 4 out of 160 patients (2.5%) suffered from congenital central hypoventilation syndrome. Only 2 of them underwent genetic testing. In 1 other, only the RET gene was analysed, and there were no abnormal findings. In the last case, the PHOX2B gene was analysed and found to be abnormal. Therefore, the prevalence of PHOX2B mutations in the overall series was 0.63%, compared to the 5% prevalence described in the medical literature. This emphasises the need to include genes other than RET in the genetic evaluation when there is clinical suspicion of a specific syndrome or associated disease.
The importance of the genetic evaluation in HSCR rests on its therapeutic and prognostic implications, not only for isolated HSCR but also for other diseases associated with HSCR. The main therapeutic and prognostic implication is that mutations in some regions of RET are associated with MEN 2A and MEN 2B syndromes,24,25 which carry an increased risk of medullary thyroid carcinoma, parathyroid hyperplasia and pheochromocytoma. Therefore, given the increased risk of developing medullary thyroid carcinoma, patients with these mutations must undergo prophylactic thyroidectomy.26 It is extremely important to know where the mutation is for the purpose of taking appropriate prophylactic measures.
The genetic study of the RET gene in HSCR is not only beneficial to patients, but also to their relatives. When a mutation is found, extension of the genetic study to first-degree relatives might reveal unknown mutations in asymptomatic individuals, who would also be candidates for prophylactic thyroidectomy and specific follow-up.
Describing mutations associated to HSCR, such as the ones found in this study, has a double implication. Firstly, it has a direct clinical implication: mutations associated to HSCR need to be known so that, when found in other patients, clinicians are aware that they are the cause of the disease and not an incidental finding, and know the exact location of the mutation in the gene, which allows knowing whether it is associated with syndromes or other diseases. Secondly, increasing the knowledge of the genetic basis of HSCR might contribute to an improved understanding of the pathogenesis of the disease in the future.
The main limitation of this study is the small number of patients that underwent genetic testing in this case series, which limited the amount of relevant genetic data found in the records. Of the 160 patients with a confirmed diagnosis of HSCR, only 21 had positive genetic testing results documented in their health records. However, we found evidence of an increasing awareness of the importance of performing genetic testing over time, with an increasing number of tests that merited reviewing and description in article, including those corresponding to the 2 novel mutations. All of the above evinces an opportunity to continue to improve by including genetic testing in the routine workup of patients with HSCR.
Conflicts of interestThe authors declare no conflicts of interest.
Please cite this article as: Lorente-Ros M, et al. Nuevas mutaciones asociadas a la enfermedad de Hirschsprung. An Pediatr (Barc). 2020;93:222–227.
