


Intracranial germ cell tumours are rare in children. They are a heterogeneous group of neoplasms that show different clinical manifestations despite having a common origin.
Patients and methodsA retrospective analysis was carried out on the epidemiological and histological characteristics, clinical manifestations, and outcomes of 20 patients diagnosed with intracranial germ cell tumours in the Niño Jesús Children's Hospital of Madrid from 1994 to 2014.
ResultsA total of 20 patients were identified: 14 boys and 6 girls. The mean age was 11.1 years (range 2–18 years). Histological confirmation of the diagnosis was obtained in 95% of the patients. Of the 20 patients, 14 were pure germinoma (70%) and 6 non-seminomatous germ cell tumours (30%). The most frequent locations were pineal (45%) and suprasellar (45%). The most frequent clinical symptoms in pineal tumours at diagnosis were headache and vomiting (77.77%), followed by visual disturbances (44.4%). In suprasellar tumours it was polydipsia and polyuria (100%). At diagnosis, 90% of the patients received radiotherapy, and 55% received chemotherapy combined with radiotherapy. There was a relapse in 4 patients (20%), and 3 of them died. Overall survival was 80%; 85.7% for pure germinomas and 60% for non-seminomatous germ cell tumours.
ConclusionsThe most common histological subtype was pure germinoma. Germ cell tumours include heterogeneous disease entities that have a variable prognosis. Thus, an accurate diagnosis is vital for patient counselling and treatment planning.
Los tumores de células germinales intracraneales son un grupo poco frecuente de tumores en niños. Comprenden un grupo heterogéneo de neoplasias, que aunque comparten un origen común, presentan comportamientos clínicos muy diferentes.
Pacientes y métodosAnálisis retrospectivo de las características epidemiológicas e histológicas, las manifestaciones clínicas y la evolución de 20 pacientes diagnosticados de tumor de células germinales intracraneal en el Hospital Infantil Universitario Niño Jesús de Madrid durante los años 1994-2014.
ResultadosSe obtuvieron 20 pacientes: 14 niños y 6 niñas. La edad media fue de 11,1 años (rango 2-18 años). Se realizó confirmación histológica en el 95% de los pacientes. De los 20 pacientes, 14 fueron germinomas puros (70%) y 6 tumores de células germinales no germinomas (30%). Las localizaciones más frecuentes fueron pineal (45%) y supraselar (45%). Los síntomas más frecuentes en el momento del diagnóstico en los tumores de localización pineal fueron cefalea y vómitos (77,77%), seguido de alteraciones visuales (44,4%), y en los tumores de localización supraselar, polidipsia y poliuria (100%). En el momento del diagnóstico recibieron radioterapia el 90% de los pacientes y quimioterapia asociada a la radioterapia el 55%. Presentaron recaída tumoral 4 pacientes (20%), de los cuales 3 fallecieron. La supervivencia global fue del 80%, siendo un 85,7% para los germinomas y un 60% para los no germinomas.
ConclusiónEl tipo histológico más frecuente fue el germinoma. Los tumores de células germinales son un grupo heterogéneo de tumores que conllevan un pronóstico diferente, por lo que un adecuado diagnóstico y estadificación es fundamental para planear el tratamiento.
Intracranial (IC) germ cell tumours (GCTs) are a rare and heterogeneous group of tumours found in adolescents and young adults. Their incidence varies widely between geographical regions, and they amount to 2–4% of brain tumours in individuals aged 0–19 years in Europe.1,2
Germ cell tumours arise from primordial germ cells that lodge ectopically in the central nervous system (CSN) during the migration from the yolk sac to the gonadal ridge and may undergo malignant transformation. Different tumours develop based on the degree of differentiation of these germ cells at this point.
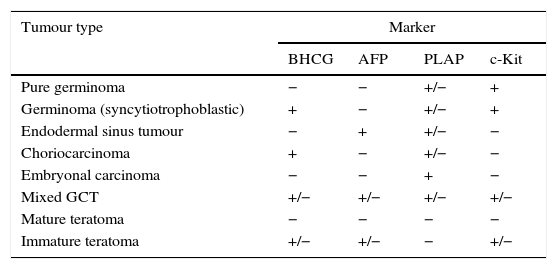
Germ cell tumours that arise in the CNS share histological, genetic and clinical characteristics with extracranial GCTs. The World Health Organization (WHO) has divided GCTs into two broad groups: germinomatous and nongerminomatous GCTs (NGGCTs)3,4 based on histology and the presence of tumour markers (Tables 1 and 2). Other classification schemes have divided GCTs into secreting and nonsecreting tumours. Secreting tumours have alpha-fetoprotein (AFP) positivity or beta-human chorionic gonadotropin (BHCG) elevations greater than 50IU/L in the cerebrospinal fluid (CSF) or any serum BHCG or AFP positivity5,6 (Table 1).
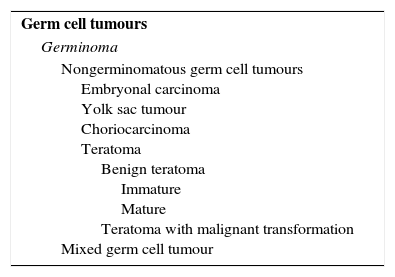
Classification of intracranial germ cell tumours proposed by the WHO.
| Germ cell tumours |
| Germinoma |
| Nongerminomatous germ cell tumours Embryonal carcinoma Yolk sac tumour Choriocarcinoma Teratoma Benign teratoma Immature Mature Teratoma with malignant transformation Mixed germ cell tumour |
From: Louis et al., editors.3
Classification of GCTs based on tumour markers.
| Tumour type | Marker | |||
|---|---|---|---|---|
| BHCG | AFP | PLAP | c-Kit | |
| Pure germinoma | − | − | +/− | + |
| Germinoma (syncytiotrophoblastic) | + | − | +/− | + |
| Endodermal sinus tumour | − | + | +/− | − |
| Choriocarcinoma | + | − | +/− | − |
| Embryonal carcinoma | − | − | + | − |
| Mixed GCT | +/− | +/− | +/− | +/− |
| Mature teratoma | − | − | − | − |
| Immature teratoma | +/− | +/− | − | +/− |
AFP, alpha-fetoprotein; BHCG, beta-human chorionic gonadotropin; PLAP, placental alkaline phosphatase.
From: Louis et al., editors.3
The clinical presentation varies based on the location and size of the tumour. The most frequent location is the pineal region, followed by the suprasellar region.7 The most common presenting symptoms are endocrine abnormalities, signs of raised intracranial pressure and visual disturbances.
Accurate staging and histological examination are important factors for the classification of patients into prognostic groups.4,8 Imaging studies cannot differentiate GCTs from other tumours, so diagnosis requires histological confirmation except in cases with elevated levels of tumour markers such as AFP and BHCG.3 Surgical management options are determined by tumour location. Traditionally, these tumours were treated with craniospinal irradiation. In the past two decades, advances in imaging techniques, radiotherapy (RTX) and surgery, and the addition of chemotherapy to treatment protocols have led to a significant improvement in the prognosis of malignant GCTs, and especially germinomas, for which the 10-year overall survival now exceeds 90%.3,9,10
Patients and methodsPatient selectionWe conducted a retrospective study of patients that received a diagnosis of IC GCT at the Department of Oncology of the Hospital Universitario Infantil Niño Jesús of Madrid between 1994 and 2014. The inclusion criteria were: (a) patients aged 18 years or less and (b) with a diagnosis of IC GCT based on the WHO classification.3 We identified 20 such patients.
We collected data for the following variables: age, sex, tumour location, symptoms at the time of diagnosis and their duration, presence of leptomeningeal metastasis, histology, tumour markers in blood and/or CSF, type of surgery performed and its complications, type of chemotherapy (CTX) and/or RTX administered and their complications, tumour recurrence, and abnormalities during long-term followup (Table 3).
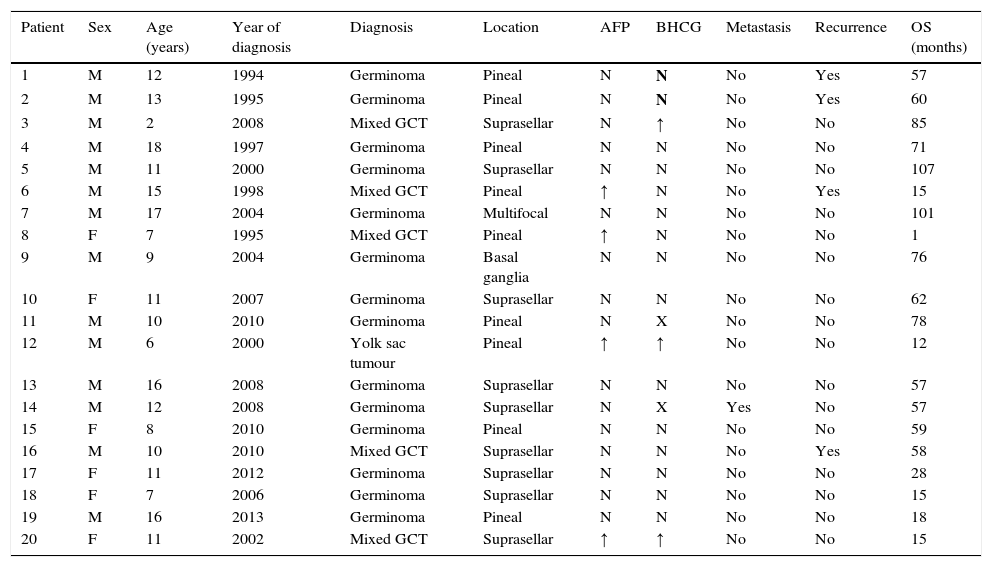
Characteristics of patients with intracranial germ cell tumours.
| Patient | Sex | Age (years) | Year of diagnosis | Diagnosis | Location | AFP | BHCG | Metastasis | Recurrence | OS (months) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 12 | 1994 | Germinoma | Pineal | N | N | No | Yes | 57 |
| 2 | M | 13 | 1995 | Germinoma | Pineal | N | N | No | Yes | 60 |
| 3 | M | 2 | 2008 | Mixed GCT | Suprasellar | N | ↑ | No | No | 85 |
| 4 | M | 18 | 1997 | Germinoma | Pineal | N | N | No | No | 71 |
| 5 | M | 11 | 2000 | Germinoma | Suprasellar | N | N | No | No | 107 |
| 6 | M | 15 | 1998 | Mixed GCT | Pineal | ↑ | N | No | Yes | 15 |
| 7 | M | 17 | 2004 | Germinoma | Multifocal | N | N | No | No | 101 |
| 8 | F | 7 | 1995 | Mixed GCT | Pineal | ↑ | N | No | No | 1 |
| 9 | M | 9 | 2004 | Germinoma | Basal ganglia | N | N | No | No | 76 |
| 10 | F | 11 | 2007 | Germinoma | Suprasellar | N | N | No | No | 62 |
| 11 | M | 10 | 2010 | Germinoma | Pineal | N | X | No | No | 78 |
| 12 | M | 6 | 2000 | Yolk sac tumour | Pineal | ↑ | ↑ | No | No | 12 |
| 13 | M | 16 | 2008 | Germinoma | Suprasellar | N | N | No | No | 57 |
| 14 | M | 12 | 2008 | Germinoma | Suprasellar | N | X | Yes | No | 57 |
| 15 | F | 8 | 2010 | Germinoma | Pineal | N | N | No | No | 59 |
| 16 | M | 10 | 2010 | Mixed GCT | Suprasellar | N | N | No | Yes | 58 |
| 17 | F | 11 | 2012 | Germinoma | Suprasellar | N | N | No | No | 28 |
| 18 | F | 7 | 2006 | Germinoma | Suprasellar | N | N | No | No | 15 |
| 19 | M | 16 | 2013 | Germinoma | Pineal | N | N | No | No | 18 |
| 20 | F | 11 | 2002 | Mixed GCT | Suprasellar | ↑ | ↑ | No | No | 15 |
AFP, alpha-fetoprotein; BHCG, beta-human chorionic gonadotropin; F, female; GCT, germ cell tumour; M, male; OS, overall survival.
The diagnosis of IC GCT was based on tumour biopsy, levels of AFP and BHCG in serum and CSF, and magnetic resonance imaging (MRI). A craniospinal MRI and examination of CSF were performed in all patients to assess for the presence of leptomeningeal metastasis. The diagnosis of NGGCT was considered in cases with histological confirmation and/or elevated levels of AFT in serum or of more than 25ng/mL in CSF and/or levels of BHCG greater than 50UI/L. All patients underwent an evaluation of pituitary-hypothalamic function at the time of diagnosis.
TreatmentSurgeryEighteen patients underwent some type of neurosurgical intervention for collection of a sample for histological confirmation. Six patients underwent an endoscopic biopsy (one transphenoidal biopsy and five through right-frontal drilling guided by a neuronavigation system), and twelve patients underwent partial resection (six occipital, three frontal and two peritoneal craniotomies, with the surgical approach unknown in the remaining patient). Eleven patients had hydrocephalus at the time of diagnosis, and eight underwent ventriculoperitoneal shunting (VPS).
ChemotherapyPatients underwent several CTX regimens based on cisplatin. Different protocols were used: UK CCSG (BEP),11 Société Française d’Oncologie Pédiatrique (SFOP),12 Children Oncology Group (COG), Pediatric Blood Cancer 200713 and SIOP CNS CGT 2003. Table 4 shows the administered drugs and their doses.
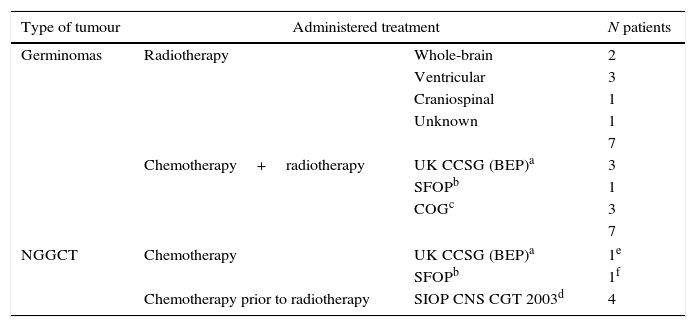
Treatment received by patients.
| Type of tumour | Administered treatment | N patients | |
|---|---|---|---|
| Germinomas | Radiotherapy | Whole-brain | 2 |
| Ventricular | 3 | ||
| Craniospinal | 1 | ||
| Unknown | 1 | ||
| 7 | |||
| Chemotherapy+radiotherapy | UK CCSG (BEP)a | 3 | |
| SFOPb | 1 | ||
| COGc | 3 | ||
| 7 | |||
| NGGCT | Chemotherapy | UK CCSG (BEP)a | 1e |
| SFOPb | 1f | ||
| Chemotherapy prior to radiotherapy | SIOP CNS CGT 2003d | 4 | |
UK CCSG (BEP): cisplatin (100mg/m2, day 1), etoposide (120mg/m2/day, days 1–3) and bleomycin (15mg/m2, day 2).11
Société Française d’Oncologie Pédiatrique: cycles 1 and 3: carboplatin (600mg/m2, day 1); etoposide (150mg/m2/day, days 1–3), cycles 2 and 4: ifosfamide (1.8g/m2/day, days 21–25) and etoposide (150mg/m2/day, days 21–23). Administration of radiotherapy after 4 cycles of chemotherapy.12
Children Oncology Group, Pediatric Blood Cancer 2007: cycle A: etoposide (100mg/m2/day, days 1–5) and cisplatin (20mg/m2/day, days 1–5). Cycle B: cyclophosphamide (1g/m2/day, days 1 and 2) and vincristine (1.5mg/m2/day, days 1, 8 and 15).13
SIOP CNS CGT 2003: PEI: cisplatin (20mg/m2/day, days 1–5), etoposide (100mg/m2/day, days 1–3), ifosfamide (1.500mg/m2/day, days 1–5).
Eighteen patients received RTX (four at the Hospital Puerta de Hierro of Madrid, 11 at the Hospital Gregorio Marañón of Madrid and 3 three in other facilities). They were treated with different RTX modalities: two with whole-brain RTX (dose, 30 and 34Gy respectively), seven with ventricular RTX (dose, 30–45Gy) and five with craniospinal RTX (dose, 23.4–30Gy). All of these patients received a boost to the tumour bed (21–59.4Gy). Three patients received only focal irradiation of the tumour (dose, 30.6–54Gy). One patient had yet to initiate RTX at the time of the analysis.
Statistical analysisWe defined overall survival as the time elapsed from diagnosis to the time of death due to all causes or the last follow-up visit. We calculated overall survival using the Kaplan–Meier method. We used the log-rank test to compare overall survival between different histological subtypes (pure germinoma compared to NGGCT). We set the baseline time point at the date of surgery for all patients.
ResultsBetween 1994 and 2014, 438 patients received a diagnosis of malignant intracranial tumour, of which 20 corresponded to IC GCTs.
The mean age at diagnosis was 11.1 years (range, 2–18 years). The incidence peaked at different ages for different histological subtypes, and we found a difference between germinomas (mean age, 12.21 years) and NGGCTs (mean age, 8.5 years). The final sample consisted of fourteen male patients and six female patients, with a higher prevalence in the male sex (2.3:1).
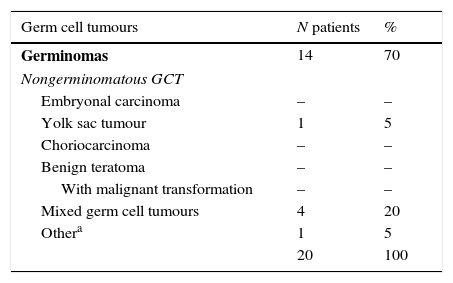
We classified the identified cases of IC GCT using the scheme proposed by the WHO.3 The most frequent histological subtype was germinoma (Table 5).
Histological subtypes found, based on the classification of germ cell tumours proposed by the WHO.
| Germ cell tumours | N patients | % |
|---|---|---|
| Germinomas | 14 | 70 |
| Nongerminomatous GCT | ||
| Embryonal carcinoma | – | – |
| Yolk sac tumour | 1 | 5 |
| Choriocarcinoma | – | – |
| Benign teratoma | – | – |
| With malignant transformation | – | – |
| Mixed germ cell tumours | 4 | 20 |
| Othera | 1 | 5 |
| 20 | 100 | |
We found elevated levels of AFP above 25ng/mL in blood or in CSF in four patients (two with mixed GCTs, one with a yolk sac tumour, and one with a secreting NGGCT for which a biopsy was not obtained) and levels of BHCG greater than 50UI/L in three out of 18 patients (one with a mixed GCT, one with a yolk sac tumour and one with a secreting NGGCT for which a biopsy was not obtained).
The location of the tumour was pineal in nine patients (45%), suprasellar in nine patients (45%), the basal ganglia and thalamus in one patient (5%) and multifocal in one other (5%).
Of all pineal tumours, 77.7% occurred in male patients. In female patients, the most frequent location was the suprasellar region (66.6%).
The median time elapsed from the onset of symptoms to diagnosis was 5 months (range, 0.1–27 months).
The most frequent symptoms at the time of diagnosis in pineal tumours were headache and vomiting (77.7%), followed by visual disturbances (44.44%): diplopia, Parinaud syndrome and erratic eye movements. The most common symptoms in patients with suprasellar tumours were polydipsia and polyuria (100%).
Of the patients with suprasellar tumours, 55.5% presented with other endocrine abnormalities at the time of diagnosis, the most frequent of which were hypothyroidism (55.5%), growth hormone deficiency (11.1%) and early puberty (22.2%).
The tumour located in the basal ganglia started with left hemiparesis, which was interpreted as ischaemic stroke on the initial MRI. The patient was a heterozygous carrier of mutation 20201 in the prothrombin gene and had abnormal levels of lipoprotein A, both of which can cause stroke in children. The patient was managed with antiplatelet therapy and follow-up care. A follow-up MRI performed seven months later detected an increase in the size of the lesion compatible with a neuroectodermal tumour, which led to performance of a biopsy.
Of all patients with suprasellar tumours, 44.4% had previously received a diagnosis of idiopathic central diabetes insipidus (DI) because the findings of the initial cranial MRI were not conclusive for diagnosing a GCT. The mean time elapsed until subsequent MRI scans detected the presence of a GCT was 21 months (range, 15–24 months).
The diagnosis was confirmed histologically in 95% of patients. The only case in which a histological assessment was not performed corresponded to a patient with a pituitary tumour that had elevated AFP and BHCG levels. In one patient, histological confirmation was not obtained at diagnosis, but was obtained when the tumour recurred.
There were no complications from the surgery in 77.7% of the patients. One patient developed an epidural haematoma following VPS that required drainage, two patients developed pneumoencephaly that resolved favourably, and one patient developed an occipital pseudomeningocoele that required drainage. There were no surgery-related deaths.
Only one patient (5%) presented with leptomeningeal spread with presence of tumour cells in the CSF at the time of diagnosis.
After the initial diagnosis, 90% of patients received RTX and 55% RTX combined with CTX. Sixty-six percent of patients tolerated RTX well. The adverse effects associated with RTX were: skin toxicity (n=1), grade 2 haematologic toxicity (n=1), grade 1 vomiting (n=1) and post-radiotherapy somnolence syndrome with headache, asthenia and somnolence that required treatment with dexamethasone (n=2).
The initial treatment included CTX in 12 patients. A total of 41 cycles of CTX were administered. The complications associated with CTX were haematologic toxicity (grades 3 and 4; n=4), gastrointestinal toxicity (grades 3 and 4; n=3), febrile neutropaenia with no documented microbial agent (n=2) and one episode of hyponatraemic seizure secondary to DI decompensation due to overhydration during the administration of CTX.
Table 4 summarises the treatments administered after histological confirmation.
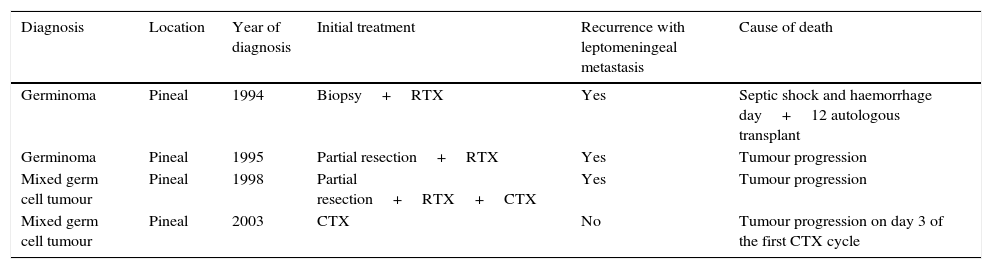
Four patients (20%) experienced relapse. Three of these patients died, and the fourth one continues in full remission after autologous stem cell transplantation. At the time of relapse, all of these patients had leptomeningeal disease and presented with clinically significant symptoms (anorexia, pain and difficulty relaxing the lower extremities, hemiparesis, lumbar pain, speech impairments and bradypsychia). In one of the four patients, the relapse occurred outside the radiation field.
The mean time to relapse was 49 months in patients with germinoma and 12.5 months in patients with NGGCTs. All relapsed patients underwent autologous stem cell transplantation following salvage CTX.
The overall survival was 84% (85.7% for patients with germinoma and 60% for patients with NGGCT), with a median duration of followup of 58 months (range, 1.43–106.9 months) (Fig. 1).
.")
One patient with a yolk sac tumour was lost to followup at 12 months from diagnosis due to transfer to another unit. Table 6 shows the data for deceased patients.
Deceased patients.
| Diagnosis | Location | Year of diagnosis | Initial treatment | Recurrence with leptomeningeal metastasis | Cause of death |
|---|---|---|---|---|---|
| Germinoma | Pineal | 1994 | Biopsy+RTX | Yes | Septic shock and haemorrhage day+12 autologous transplant |
| Germinoma | Pineal | 1995 | Partial resection+RTX | Yes | Tumour progression |
| Mixed germ cell tumour | Pineal | 1998 | Partial resection+RTX+CTX | Yes | Tumour progression |
| Mixed germ cell tumour | Pineal | 2003 | CTX | No | Tumour progression on day 3 of the first CTX cycle |
CTX, chemotherapy; RTX, radiotherapy.
The side effects associated with the disease and the treatment observed during long-term followup were: hypothalamic-pituitary hormone imbalances that required hormone replacement therapy (45%), difficulty concentrating and memory impairment (20%), chronic kidney disease (10%), papillary thyroid carcinoma (5%) and bitemporal hemianopsia (5%).
DiscussionIntracranial GCTs are rare in children. In our study, they amounted to 4.5% of all malignant intracranial tumours diagnosed between 1994 and 2004 at the Hospital Niño Jesús. This percentage is similar to those reported by other case series in the United States and Europe.4
The prevalence is higher in males,14–16 a predominance that is even more marked in NGGCTs.14,17 The most frequent tumour locations are pineal and suprasellar.7 In our series, consistent with the literature,14,18 suprasellar locations predominated in female patients.
Tumours in the basal ganglia are rare, corresponding to 4–15% of patients with IC GCTs, but it is worth discussing them due to their clinical characteristics and the challenges they pose to diagnosis. Hemiparesis is the most frequent symptom.1,19 In our series, one patient with a tumour located in the basal ganglia had onset with left hemiparesis, which was interpreted based on the findings of the initial MRI as a stroke due to the characteristics of the patient (carrier of a mutation in the prothrombin gene and abnormal lipoprotein A levels). Follow-up imaging detected the tumour and led to diagnosis.
The symptoms in our series were similar to those described by other authors. Pineal tumours usually present with symptoms of raised intracranial pressure.7,15 Ophthalmological abnormalities are found in 25–50% of patients, and Parinaud syndrome in up to 72.4%.4,7,15 We must make a point of including pineal tumours in the differential diagnosis of patients presenting with symptoms of raised intracranial pressure or ocular symptoms.
Suprasellar tumours usually present with DI, delayed sexual development, hypothyroidism and early puberty.15,20,21 They may also present with ophthalmological abnormalities. In our series, 44.4% of patients with suprasellar GCTs received an initial diagnosis of idiopathic central DI because the tumour was not detectable in the initial MRI. In these patients, the mean time elapsed from the onset of symptoms with normal MRI findings to the evidence of suprasellar tumour in MRI was 21 months, which was consistent with previous reports in the literature.7,22,23 Patients with IC GCTs frequently have endocrine symptoms that may last for a long time before the tumour can be verified by imaging. The possibility of a GCT should be considered in patients presenting with symptoms of endocrinopathy, and these patients should undergo imaging tests periodically, as delayed diagnosis increases the risk of disseminated disease.24
In our series, similar to what other studies have reported,5 45% of patients needed hormone replacement therapy after completing treatment, which requires long-term endocrine followup.
Germ cell tumours can be divided into two broad categories: germinomas and nongerminomatous germ cell tumours. Germinomas account for approximately 50–70% of GCTs.4,20 This distinction is very relevant in clinical practice, as histological subtype is the most significant isolated prognostic factor in IC GCTs.7,14,16
Different authors have proposed the therapeutic classification of IC GCTs based on histology15,25 categorising IC GCTs as having a good, intermediate, or poor prognosis (Table 7).
Therapeutic classification of intracranial germ cell tumours.
| Group with good prognosis |
| Pure germinoma |
| Mature teratoma |
| Group with intermediate prognosis |
| Germinoma with syncytiotrophoblastic giant cells |
| Immature teratoma |
| Teratoma with malignant transformation |
| Mixed tumours mainly composed of germinoma or teratoma |
| Group with poor prognosis |
| Choriocarcinoma |
| Yolk sac tumour |
| Embryonal carcinoma |
| Mixed tumours mainly composed of choriocarcinoma, yolk sac tumour, or embryonal carcinoma. |
Source: Matsutani et al.15
Germinomas are extremely sensitive to CTX and radiation. In the past 20 years, craniospinal irradiation has been the gold standard of treatment and has achieved 5-year survival rates of more than 90%.4,6,10,26 However, this approach is associated with adverse effects on the CNS, thyroid function and growth and development.5,27–29 In patients with NGGCTs, despite the use of intensified CTX and high-dose RTX, 10-year survival continues to be as low as 30–80%.22,23,30 Currently, multicentric clinical trials (International Society of Pediatric Oncology CTG phase ii) are underway that aim at determining the minimum dose and radiation volume that can be administered without jeopardising survival in localised germinomas4,14,15 and, for NGGCT, the dosages needed improve survival in high-risk patients by intensifying treatment, especially in children that receive the diagnosis at age less than 6 years and patients with AFP levels greater than 1000ng/mL.
In our series, the survival in the germinoma group was lower than that reported in the literature. Two out of fourteen patients with pure germinoma died, but we ought to highlight that these patients received the diagnosis in 1994 and 1995, respectively. None of the patients that received a diagnosis of germinoma between 1996 and 2014 died. In our study, the difference in overall survival between patients with germinoma and patients with NGGCT was not statistically significant, but this was probably due to the small sample size.
In our sample, the diagnosis was confirmed histologically in 95% of patients. The only case in which histological assessment was not performed corresponded to a patient with a pituitary tumour that had elevated AFP and BHCG levels. Given the prognosis of this group of tumours, making an accurate diagnosis with histological confirmation is essential to plan an appropriate treatment strategy. Neuroimaging tests cannot differentiate GCTs from other tumours, and therefore diagnosis requires histological confirmation7 except in cases in which there is elevation of tumour markers.
In the past, the risks associated with surgery led to the use of therapeutic irradiation trials to confirm the diagnosis of germinoma. At present, advances in neurosurgical technique have made resection of pineal tumours much safer, with a mortality rate of less than 5%.15,31
Given the favourable response of germinomas to combined CTX and RTX, the extent of resection in these cases is limited by the preservation of visual and neuroendocrine function.5 However, more aggressive approaches have a greater impact on NGGCTs, and higher survival rates have been reported in patients that have undergone radical surgery.5,15,32 In cases of mature teratoma, extended resection is usually curative.
Accurate staging is essential for treatment planning.4,33 There is no universally accepted staging system for germinoma, and most researchers use the TNM system used for medulloblastomas.23 The following workup should be performed in all patients for diagnosis: craniospinal MRI, measurement of AFP and BHCG in serum and CSF, lumbar puncture for CSF cytology, evaluation of pituitary function, visual field test and neuropsychological evaluation.4,5,34
In our study, we found relapses in two out of the fourteen patients with germinoma (14.2%). Both had been treated with whole-brain RTX with boosts of the tumour bed. In patients with NGGCT, the relapse rate was 33.3%. The mean time to relapse was 49 months in patients with germinoma and 12.5 months in patients with NGGCT. Previous studies have reported relapse rates of 10% for germinomas5,35 and of up to 55.7% for NGGCTs.15 The mean time elapsed to relapse in germinomas ranges between 30 and 50 months.1,36 Late relapses after 8–15 years are not uncommon, so patients need to remain under observation for a period of at least 10 years.1
In the four patients that relapsed in our series, the recurrence occurred with leptomeningeal spread. According to the literature, the most common type of relapse in IC GCTs is local recurrence at the primary site, but leptomeningeal spread is found in 30%. The prognosis of relapsing patients, especially of those with NGGCT, continues to be poor.4,37 Salvage treatment includes CTX, RTX and myeloablative CTX with autologous stem cell transplantation.38,39
ConclusionIntracranial germ cell tumours are a heterogeneous group of tumours that have different prognoses, and therefore accurate diagnosis and staging is essential for treatment planning. Since the number of patients that recover from these tumours is increasing over time, we should identify low-risk patients in whom treatment could be minimised to reduce its impact on the quality of life of survivors without jeopardising long-term survival, and intensify treatment in high-risk groups to achieve a higher rate of recovery.1,5
Conflicts of interestThe authors have no conflict of interests to declare.
Please cite this article as: Cormenzana Carpio M, Nehme Álvarez D, Hernández Marqúes C, Pérez Martínez A, Lassaletta Atienza A, Madero López L. Tumores germinales intracraneales: revisión de 21 años. An Pediatr (Barc). 2017;86:20–27.
