



Pontocerebellar Hypoplasia (PCH) refers to a heterogeneous group of rare neurodegenerative disorders with autosomal recessive inheritance characterised by hypoplasia of cerebellum and pons associated with progressive microcephaly.1 Ten subtypes have been identified to date (PCH1-10); of which the individual incidence is unknown and the genotype–phenotype correlation has yet to be fully determined. Pontocerebellar hypoplasia type 1 (PCH1), the most prevalent, is distinctively associated with neuronal degeneration in the anterior horn of the spinal cord, giving rise to a clinical picture compatible with spinal muscular atrophy type 1 that includes severe hypotonia and significant feeding difficulties.2,3 In this article, we present the cases of two siblings, born to healthy nonconsanguineous parents and of Romani descent, that had PCH type 1B associated with a mutation in the EXOSC gene. There was no relevant family history.
Case 1Female newborn. Pregnancy with few checkups and normal prenatal ultrasound findings. Uncomplicated term delivery with Apgar score of 7/8. The patient required resuscitation with positive pressure ventilation in the delivery room. The physical examination revealed generalised severe hypotonia with very reduced deep tendon reflexes, shallow breathing, arthrogryposis with contractures in hands and elbows, genu recurvatum in the right lower extremity and fracture at the epiphyseal-metaphyseal junction in the distal third of the femur. The facial phenotype was not dysmorphic, save for the features resulting from hypotonia. Anthropometric measurements at birth: weight, 2330g (5th–10th percentile); length, 44.5cm (<3rd percentile); head circumference, 32.5cm (10th–25th percentile). The patient needed mechanical ventilation due to progressive respiratory failure and placement of a nasogastric tube for enteral nutrition due to inefficient sucking and swallowing. Peripheral hypotonia was suspected, leading to performance of electromyography (EMG), which revealed a clear neuropathic pattern. The karyotype was 46, XX. The results of single-gene testing of the SMN1 gene were normal, ruling out spinal muscular atrophy type 1. The unexpected finding of hypoplasia of the pons, vermis and cerebellar hemispheres in magnetic resonance imaging (MRI) of the head, along with ventriculomegaly with normal intracranial pressure, guided the diagnosis of PCH type 1, which was subsequently confirmed by molecular testing, which identified a homozygous missense mutation in exon 1 of the EXOSC gene (c.92G>C). The patient died at age 4 months of complications of aspiration pneumonia.
Case 2Male. Uncomplicated delivery at 39 weeks’ gestation, with normal findings in prenatal ultrasound examinations. Apgar score of 8/9. The patient did not need resuscitation in the delivery room. Anthropometric measurements at birth: weight, 2560g (<5th percentile); length, 46.7cm (10th–25th percentile); head circumference, 32.5cm (<5th percentile). The clinical presentation was similar to that of his deceased sister, requiring mechanical ventilation and enteral feeding through a nasogastric tube. The karyotype was 46, XY. Head MRI revealed hypoplasia of the pons and cerebellum, compatible with the diagnosis of PCH type 1 (Fig. 1). The patient died at age 4½ months due to cardiorespiratory failure. The findings of the postmortem histopathological examination (spinal cord and muscle) were consistent with spinal muscular atrophy, with gliosis and a reduced number of Purkinje cells in the granular layer in the cerebellum. Molecular testing detected the same homozygous mutation (c.92G>C) in the EXOSC3 gene, confirming the clinical diagnosis. Both parents were heterozygous carriers of the mutation.
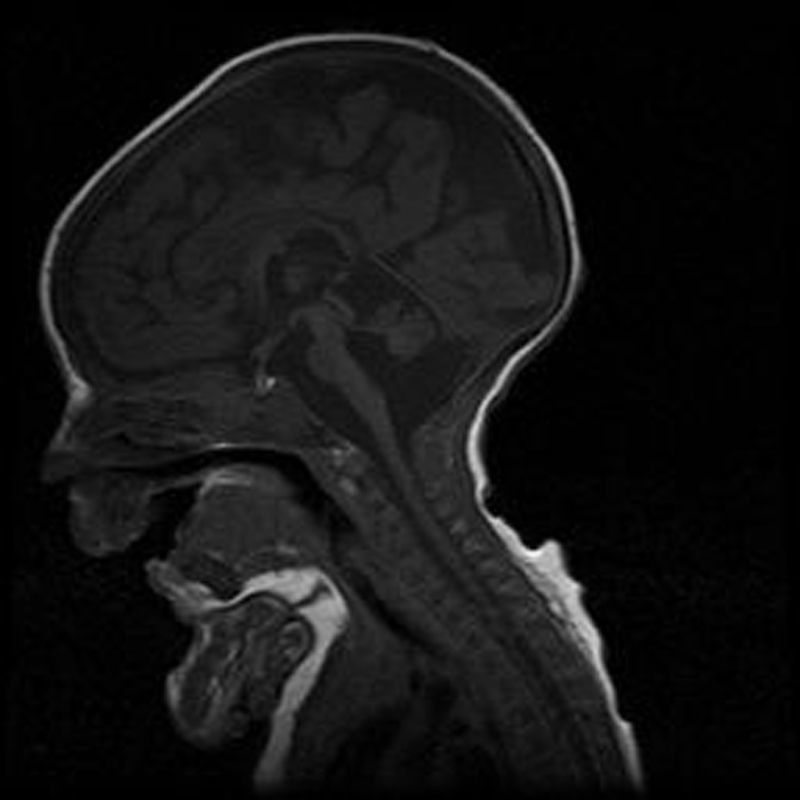
Pontocerebellar hypoplasia type 1 (OMIM# 606489) manifests in the neonatal period with severe hypotonia, absent or reduced deep tendon reflexes, contractures and very weak sucking requiring the use of nasogastric feeding to ensure adequate nutrition. It is associated with a high incidence of aspiration pneumonia, whose complications are usually the cause of early death. Table 1 summarises the clinical criteria for the diagnosis of PCH type 1. The prognosis is highly uncertain, and symptomatic treatment and support measures are the only options currently available. Survival varies, with ranging from infancy to adolescence,4 and there are no biological or genetic markers that allow reliable prognostication. Most sporadic cases of PCH had initially been associated with mutations in various genes (TSEN54, RARS2 and VRK) until the identification of the EXOSC35 gene, of which mutations have been detected in 50% of the cases studied. The EXOSC3 gene encodes a component of the human exosome, a protein complex involved in RNA processing. Its discovery provided evidence than changes in the exosome can cause disease in humans.5 The c.92G>C mutation in the EXOSC3 gene has been identified in some cases of PCH1 reported in the literature, especially in the Czech Romani population, and associated with the most severe form of disease.6
Diagnostic criteria for PHC type 1.
| Criteria | Major | Minor |
|---|---|---|
| Clinical-neurologic | Hypotonia Muscle atrophy Dystonia Spasticity EMG: lower motor neuron involvement | Contractures Swallowing insufficiency Nystagmus Seizures Strabismus |
| Neuroradiologic | Hypoplasia and/or atrophy of the cerebellum Hypoplasia and/or atrophy of the pons Cerebellar vermis and cerebellar hemispheres equally affected | Intracerebellar cysts Ventriculomegaly |
| Neuropathologic | Muscle | Spinal cord | Cerebellum |
|---|---|---|---|
| Neurogenic muscle atrophy | Degeneration and loss of motor neurons in the anterior spinal horn | Loss of Purkinje cells. Folial atrophy. Degeneration of dentate nuclei. |
Source: Eggens et al.4
We ought to mention that the EXOSC3 gene is not included in many of the gene panels designed for the assessment of PCH, which should be taken into account in the investigation of these patients given the large size of the Romani population in Spain.
Please cite this article as: Di Giovambattista AP, Jácome Querejeta I, Ventura Faci P, Rodríguez Martínez G, Ramos Fuentes F. Hipoplasia pontocerebelosa tipo I familiar con mutación en EXOSC3. An Pediatr (Barc). 2017;86:284–286.
Previous presentation: This study was presented as a poster with a short oral communication at the 25 Congreso de Neonatología y Medicina Perinatal, May 20–22, 2015; Seville, Spain.

Anales de Pediatría (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals