



Las hipoplasias pontocerebelosas (HPC) son un grupo heterogéneo de trastornos neurodegenerativos raros con herencia autosómica recesiva, caracterizados por hipoplasia de cerebelo y protuberancia, asociada a microcefalia progresiva1. Actualmente se conocen 10 subtipos (HPC1-10) cuya incidencia individual es desconocida, y en las que la correlación genotipo-fenotipo es todavía objeto de estudio. La HPC1, la más prevalente, presenta la peculiaridad de asociar una degeneración de las neuronas del asta anterior de la médula, dando lugar a una clínica superponible a una atrofia muscular espinal tipo I, y que incluye hipotonía grave y dificultades importantes para la alimentación2,3. En este trabajo presentamos el caso de 2 hermanos de progenitores sanos no consanguíneos, de origen rumano, afectados por HPC tipo 1B asociada a mutación en el gen EXOSC. No constataban antecedentes familiares de interés.
Caso 1Mujer. Embarazo poco controlado, con ecografías prenatales normales. Parto eutócico a término con Apgar 7/8. Precisó reanimación con oxígeno a presión positiva en paritorio. A la exploración física destacaba una hipotonía generalizada grave con reflejos osteotendinosos muy disminuidos, respiración superficial, artrogriposis con contracturas en manos y codos, genu recurvatum en la extremidad inferior derecha y fractura epifisio-metafisaria en el tercio distal del fémur. Fenotipo facial no dismórfico, salvo los rasgos derivados de su hipotonía. Antropometría neonatal: P 2.330g (p5-10), L 44,5cm (p<3), PC 32,5cm (p10-25). Necesitó ventilación mecánica por insuficiencia respiratoria progresiva, así como nutrición enteral por sonda nasogástrica, dada la succión y deglución ineficaz. Ante la sospecha clínica de hipotonía de origen periférico se solicitó un EMG, que mostró un claro patrón neuropático. Cariotipo 46, XX. Se realizó el estudio del gen SMN1 que fue negativo y se descartó la atrofia muscular espinal tipo I. El hallazgo inesperado de una hipoplasia del puente, del vermis y de los hemisferios cerebelosos en la RM cerebral, junto a una ventriculomegalia, no a tensión, orientó al diagnóstico de HPC tipo 1, confirmada posteriormente con el estudio molecular, que identificó una mutación tipo cambio de sentido (missense) en homocigosis en el exón 1 del gen EXOSC (c.92G>C). La paciente falleció a los 4 meses de vida por complicaciones derivadas de una neumonía por aspiración.
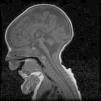
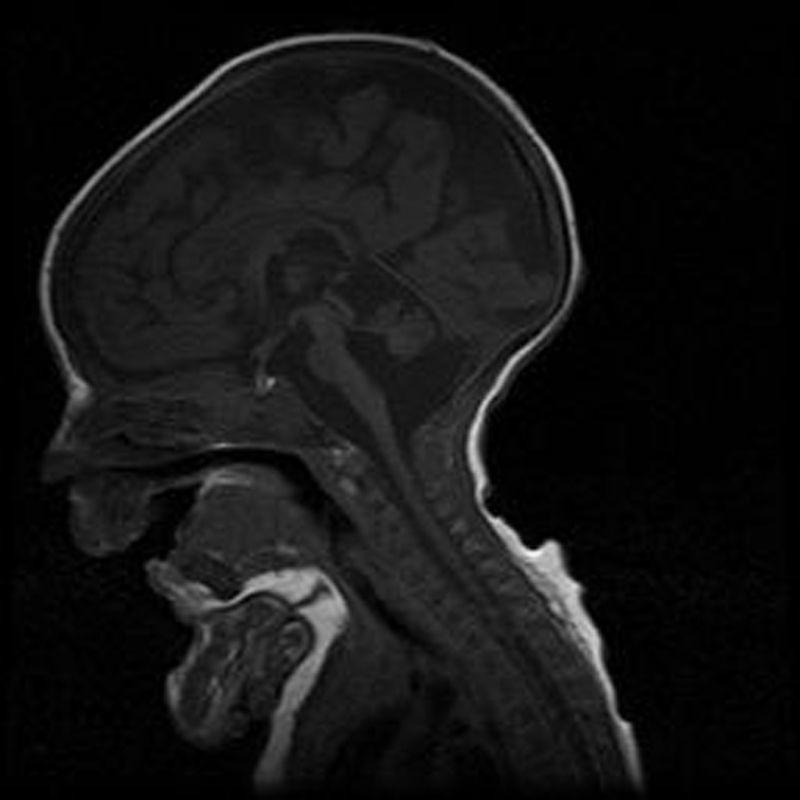
Caso 2Varón. Parto eutócico tras 39 semanas de embarazo, con ecografías prenatales normales. Apgar 8/9. No precisó reanimación en paritorio. Antropometría neonatal: P 2.560g (<p5), L 46,7cm (p10-25), PC 32,5cm (<p5). El cuadro clínico fue similar al de su hermana fallecida, precisando ventilación mecánica y alimentación enteral por SNG. Cariotipo 46, XY. La RM cerebral evidenció la presencia de hipoplasia de puente y cerebelo, que era compatible con el diagnóstico de HPC tipo 1 (fig. 1). Falleció a los 41/2 meses de vida por fracaso cardiorrespiratorio. Los estudios anatomopatológicos post mortem (médula espinal y músculo) fueron compatibles con atrofia muscular espinal y a nivel de cerebelo se encontró disminución de las células de Purkinje, de la capa granulosa y gliosis. El estudio molecular identificó la misma mutación (c.92G>C) en homocigosis en el gen EXOSC3, lo que confirmó el diagnóstico clínico. Ambos padres eran portadores heterocigotos de la mutación.
La HPC tipo 1 (OMIM# 606489) se presenta ya en el periodo neonatal con hipotonía grave, reflejos osteotendinosos ausentes o disminuidos, contracturas y succión muy débil, que requiere la utilización de SNG para asegurar la nutrición. Es frecuente la aparición de neumonías por aspiración, cuyas complicaciones suelen ser la causa del fallecimiento precoz. Los criterios clínicos necesarios para el diagnóstico de HPC tipo 1 se resumen en la tabla 1. El pronóstico es muy incierto ya que el tratamiento disponible es únicamente sintomático y de soporte. La supervivencia es variable, desde la infancia hasta la adolescencia4, no existiendo marcadores biológicos ni genéticos que permitan adelantar su pronóstico de forma fiable. La mayoría de los casos esporádicos de HPC estaban asociados a mutaciones en varios genes (TSEN54, RARS2 y VRK), hasta que se produjo el descubrimiento del gen EXOSC35, donde se identificaron mutaciones en alrededor del 50% de los casos estudiados. El gen EXOSC3 codifica un componente del exosoma, que es un complejo multiprotéico involucrado en el procesamiento del ARN. Gracias a su descubrimiento se pudo demostrar que las anomalías en el exosoma pueden causar enfermedades en la especie humana5. La mutación c.92G>C en EXOSC3 ha sido identificada en algunos casos de HPC tipo 1 publicados, en particular en la población checa-romaní, y está asociado a la forma más grave de la enfermedad6.
Criterios diagnósticos de la HPC tipo i
| Criterios | Mayores | Menores | |
|---|---|---|---|
| Clínico-neurológicos | Hipotonía Atrofia muscular Distonía Espasticidad EMG: patrón anómalo en motoneurona inferior | Contracturas Alteración de la deglución Nistagmo Convulsiones Atrofia óptica | |
| Neuro-radiológicos | Hipoplasia y/o atrofia de cerebelo Hipoplasia y/o atrofia de puente Afectación igual de vermis y hemisferios cerebelosos | Quiste intracerebeloso Ventriculomegalia | |
| Anatomopatológicos | Músculo | Médula | Cerebelo |
| Atrofia muscular neurogénica | Degeneración de las motoneuronas del asta anterior de la médula | Pérdida de las células de Purkinje. Atrofia del folium cerebeloso. Degeneración del núcleo dentado |
Fuente: Eggens et al.4.
Cabe señalar que el gen EXOSC3 no aparece en muchos de los paneles genéticos diseñados para el estudio de HPC, lo que ha de ser tenido en cuenta en el estudio de estos pacientes, dada la amplia población romaní que hay en nuestro país.
Trabajo presentado en calidad de póster con defensa, en el 25 Congreso de Neonatología y Medicina Perinatal, Sevilla, 20-22 de mayo de 2015.
