
Coenzyme Q10 (CoQ10) plays a crucial role in several cellular processes, such as such as energy production through the mitochondrial respiratory chain, β-oxidation of fatty acids and pyrimidine biosynthesis, and it is also one of the main cellular antioxidants. Its synthesis requires multiple enzymes encoded by different genes (PDSS1, PDSS2, COQ2, COQ4, COQ6, COQ8A, COQ8B and COQ9). Due to the ubiquity of CoQ10 and the fact that the tissue expression of enzymes needed for its synthesis is variable, the clinical spectrum of CoQ10 deficiency is very heterogeneous, with the disorder potentially manifesting with myopathy, psychomotor retardation (PMR), encephalopathy, cerebellar ataxia, retinopathy, pulmonary hypertension, myocardiopathy, steroid-resistant nephrotic syndrome (SRNS) and chronic kidney disease (CKD), depending on the involved gene.1
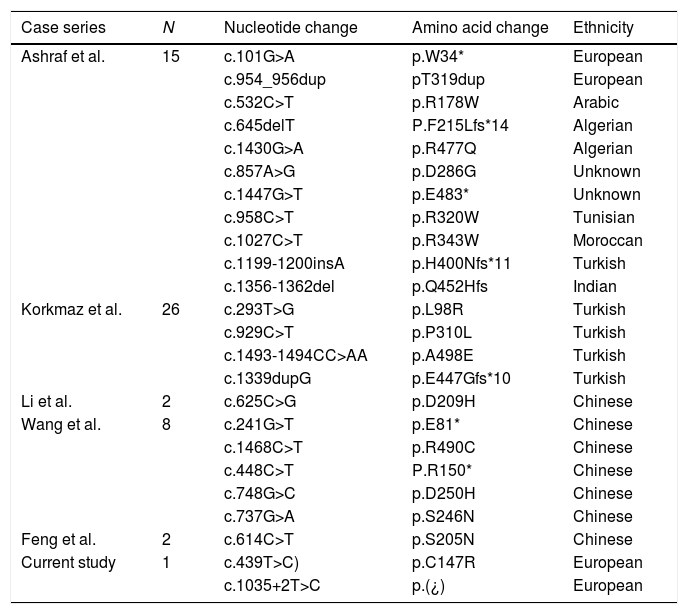
We present the case of a boy aged 6 years with a past history of febrile seizures and PMR of unknown aetiology admitted to hospital with hypertensive encephalopathy (blood pressure, 240/170mmHg). The parents reported the child had experienced asthenia, polyuria, polydipsia and foamy urine lasting 6 months and palpebral oedema in the past few days. A head computed tomography (CT) scan was performed, with normal findings. The evaluation revealed the presence of microscopic haematuria, nephrotic-range proteinuria (urine protein/creatinine ratio, 23mg/mg; normal range <0.2) and kidney failure with an estimated glomerular filtration rate of 4mL/min/1.73m2 (KDIGO 2012 stage 3). The ultrasound scan revealed a small renal size for age, loss of corticomedullary differentiation and increased echogenicity, findings suggestive of long-term renal disease. There was also evidence bilateral retinitis pigmentosa. Continuous venovenous haemodiafiltration was initiated to control hypervolemia and manage electrolyte abnormalities such as hyperphosphataemia and hyperkalaemia, and chronic haemodialysis was maintained for 3 months until the patient underwent transplantation of a kidney from a deceased donor, which was successful. Genetic testing for nephronophthisis (NPHP1–4) was negative. Later on, the evaluation was expanded with next-generation sequencing of 140 genes associated with renal diseases, which identified 2 novel compound heterozygous variants of gene COQ8B (ADCK4) (c.439T>C [p.Cys147Arg] and c.1035+2T>C [p.?]), one of the genes involved in the synthesis of CoQ10 (Table 1).
Previously described COQ8B variants and published case series.
| Case series | N | Nucleotide change | Amino acid change | Ethnicity |
|---|---|---|---|---|
| Ashraf et al. | 15 | c.101G>A | p.W34* | European |
| c.954_956dup | pT319dup | European | ||
| c.532C>T | p.R178W | Arabic | ||
| c.645delT | P.F215Lfs*14 | Algerian | ||
| c.1430G>A | p.R477Q | Algerian | ||
| c.857A>G | p.D286G | Unknown | ||
| c.1447G>T | p.E483* | Unknown | ||
| c.958C>T | p.R320W | Tunisian | ||
| c.1027C>T | p.R343W | Moroccan | ||
| c.1199-1200insA | p.H400Nfs*11 | Turkish | ||
| c.1356-1362del | p.Q452Hfs | Indian | ||
| Korkmaz et al. | 26 | c.293T>G | p.L98R | Turkish |
| c.929C>T | p.P310L | Turkish | ||
| c.1493-1494CC>AA | p.A498E | Turkish | ||
| c.1339dupG | p.E447Gfs*10 | Turkish | ||
| Li et al. | 2 | c.625C>G | p.D209H | Chinese |
| Wang et al. | 8 | c.241G>T | p.E81* | Chinese |
| c.1468C>T | p.R490C | Chinese | ||
| c.448C>T | P.R150* | Chinese | ||
| c.748G>C | p.D250H | Chinese | ||
| c.737G>A | p.S246N | Chinese | ||
| Feng et al. | 2 | c.614C>T | p.S205N | Chinese |
| Current study | 1 | c.439T>C) | p.C147R | European |
| c.1035+2T>C | p.(¿) | European |
On account of the genetic diagnosis, the patient started treatment with ubiquinone (5mg/kg/day), which improved psychomotor development and academic performance. At present, the patient is aged 12 years, the renal graft remains functional and his blood pressure is normal. The patient has not experienced any more seizures and treatment with anticonvulsants was discontinued successfully. In the physical examination, the patient continues to exhibit mild dysmetria in the finger-to-nose test and intention tremor in the hands despite normal findings of magnetic resonance imaging at age 11 years.
In CoQ10 deficiency, glomerular involvement, whether isolated or syndromic, is associated with changes in genes COQ2, COQ6, PDSS2 and COQ8B (ADCK4). Between 1% and 1.9% of cases of SRNS are due to CoQ10 deficiency associated with variants of these genes.
COQ8B encodes a kinase (aarF domain-containing kinase 4, ADCK4)2 expressed in mitochondria and in particular in pedicels, the proximal tubules and the collecting ducts. It also interacts with COQ6 at the podocyte level.3
Changes in the COQ8B gene lead to loss of function in ACDK4, resulting in decreased levels of CoQ10 and decreased podocyte migration in absence of proliferation or apoptosis of fibroblasts or in podocytes. At onset, which typically occurs in adolescence, 44% of patients present with SRNS and 46% with advanced CKD (stage 3–5).4 The renal histology is focal segmental glomerulosclerosis. Some patients also present with extrarenal manifestations, such as retinitis pigmentosa, convulsive seizures or PMR. Patients that have onset with SRNS may rapidly progress to terminal CKD and require a kidney transplant.
The administration of CoQ10 can improve symptoms, lower protein levels in urine and slow down the progression of CKD. Case series of CoQ10 deficiency caused by COQ8B variants (ADCK4) published to date have shown that 4 out of 43 patients had a partial or total response to this treatment.3–5 In 3 of these patients, renal disease was in the early stages.
Genetic testing including investigation of genes involved in the biosynthesis of CoQ10 should be considered in patients presenting with SRNS or CKD of unknown aetiology with or without extrarenal manifestations such as retinitis pigmentosa, convulsive seizures or PMR. It offers multiple benefits: an unequivocal molecular diagnosis for patients and their families, knowledge of the genotype-phenotype association and delivery of treatment which, if initiated early, may partially or completely halt the progression of renal and extrarenal disease. Genetic testing through multi-gene next generation sequencing panels with subsequent confirmation by Sanger sequencing is the most cost-effective approach at the moment.6
The case presented here illustrates the added value of genetic diagnosis in paediatric patients with severe CKD of unknown aetiology and the importance of identifying any associated abnormalities.
Please cite this article as: AdánLanceta V, Romero Salas Y, Justa Roldán ML, García Jiménez MC, Ariceta Iraola G. Encefalopatía, fallo renal y retinopatía. Déficit de CoQ10 pormutación de COQ8B. An Pediatr (Barc). 2021;94:415–417.
