

Congenital hyperinsulinism (CH) is a severe disorder characterised by the appearance of severe hypoglycaemia. Pathogenic mutations in the ABCC8 and KCNJ11 genes are the most frequent cause, although its appearance also been associated to mutations in other genes (GCK, GLUD1, HADH, HNF1A, HNF4A, SLC16A1, UCP2, HK1), and with different syndromes.
Materials and methodsRetrospective review of patients diagnosed with CH in this unit during the last 18 years (2001–2018). Genetic analysis included screening for 11 genes in genomic DNA from peripheral blood (ABCC8, GCK, GLUD1, HADH, HNF1A, HNF4A, INSR, KCNJ11, SLC16A1, UCP2, and SLC25A15).
ObjectiveTo carry out a clinical and genetic characterisation of the diagnosed cases of CH in Gran Canaria.
ResultsThere have been 10 cases of persistent HC since 2001. Seven of them had mutations in the ABCC8 gene, one in the HNF4α gene, and in two patients, no pathogenic mutations were found in the analysed genes. Four patients presented with previously undescribed mutations. Pancreatectomy was performed in two of the cases. The minimum insulin value detected in hypoglycaemia was 6.81 µIU/mL. The incidence of persistent CH for Gran Canaria and Lanzarote is 1/15,614.
ConclusionsFour patients had previously undescribed mutations. The most frequently affected gene was ABCC8. Pancreatectomy was required in 20% of the patients. An insulin value of ≥6.81 µIU/mL was observed in all patients at the time of diagnosis. The incidence of CH in Gran Canaria is high.
El hiperinsulinismo congénito (HC) es una patología severa caracterizada por la aparición de hipoglucemias graves. Las mutaciones patogénicas en los genes ABCC8 y KCNJ11 son la causa más frecuente, aunque también se han descrito en otros (GCK, GLUD1, HADH, HNF1A, HNF4A, SLC16A1, UCP2, HK1) y asociado a diferentes síndromes.
Material, pacientes y métodosRevisión retrospectiva de los pacientes con diagnóstico de HC en nuestra unidad durante los últimos 18 años (2001–2018). El análisis genético incluyó un cribado de 11 genes en ADN genómico a partir de sangre periférica (ABCC8, GCK, GLUD1, HADH, HNF1A, HNF4A, INSR, KCNJ11, SLC16A1, UCP2, SLC25A15).
ObjetivosRealizar una caracterización clínica y genética de los casos diagnosticados de HC en nuestro medio.
ResultadosDesde 2001 hemos tenido 10 casos de HC persistente. Siete presentaron mutaciones en el gen ABCC8, uno en el gen HNF4α y en dos pacientes no se encontraron mutaciones patogénicas en los genes analizados. Cuatro pacientes presentaron mutaciones no descritas previamente. Se recurrió a la pancreatectomía en 2 de los casos. El valor mínimo de insulina detectado en hipoglucemia fue de 6,81 µUI/mL. La incidencia de HC persistente para Gran Canaria y Lanzarote es de 1/15.614.
ConclusionesCuatro pacientes presentaron mutaciones no descritas. El gen más frecuentemente afectado fue ABCC8. Un 20% de los pacientes requirieron pancreatectomía. En todos los pacientes se objetivó un valor de insulina ≥6,81 µUI/mL en el momento del diagnóstico. La incidencia de HC en Gran Canaria es elevada.
Congenital hyperinsulinism (CHI) is a rare disease with a variable incidence, with previously reported frequencies including 1 per 50 000 live births (1990, Netherlands),1 1 in 35 400 (2011, Japan)2 and 1 in 2500 (1998, Saudi Arabia).3
It is a severe and potentially fatal disease characterised by uncontrolled insulin secretion leading to frequent episodes of hypoglycaemia. Since these episodes can affect the neurologic development of affected patients, early diagnosis and initiation of appropriate treatment is essential. The age of onset of hypoglycaemia varies, ranging from days to years after birth.
The diagnosis of CHI may be challenging. In patients with recurrent hypoglycaemia, obtention of a critical sample during the episode of hypoglycaemia is recommended to assess the levels of the main regulators of blood glucose levels (a glucose level of less than 50 mg/dL is considered low enough to merit investigation). If an episode of hypoglycaemia does not develop spontaneously, attempts should be made to provoke one through prolonged fasting with a specified duration based on the age of the patient (12–18 h in the neonatal period, 24 h in infants, 36 h in children aged more than 1 year and up to 72 h in adolescents and adults).4 The presence of at least 2 of the following criteria is considered diagnostic of CHI in the context of hypoglycaemia5: detectable insulin levels, increase in blood glucose level after administration of glucagon (>30 mg/dL) and need of high doses of intravenous glucose to maintain blood glucose in the normal range (3–7 mg/k/min in infants aged less than 6 months and more than 7 mg/k/min in children aged more than 6 months). Other findings also support the diagnosis, such as low levels of ketone bodies (β-hydroxybutyrate), free fatty acids or insulin-like growth factor binding protein 1 (IGFBP1).6
Variants in the genes encoding pancreatic β cell potassium channels (ABCC8 and KCNJ11 genes) are the most frequent cause (accounting for 40%–60% of cases), although variants in other genes have been described either in isolation (GCK, GLUD1, HADH, HNF1α, HNF4α, SLC16A1, UCP2, HK1), associated with other abnormalities (PGM1, PMM2, CACNA1D, FOXA2) or in the context of known syndromes (Beckwith–Wiedemann, Sotos and Kabuki syndromes, among others).7,8 Notwithstanding, an aetiological diagnosis is not made in 30%–55% of cases of persistent CHI.7,9
From a histological perspective, pancreatic involvement may be diffuse or localised. In the former, abnormal β cells are found throughout the pancreatic tissue, while the latter corresponds to a focal pancreatic adenomatous hyperplasia.10 Differentiating these two forms requires positron emission tomography imaging with levodopa (PET-L-DOPA).
There is a clear genotype-phenotype correlation in the response to treatment in CHI. Patients with potassium channel variants exhibit a poorer response to pharmacotherapy. The presence of biallelic (homozygous or compound heterozygous) pathogenic variants blocks the expression of ATP-sensitive potassium channels (KATP), resulting in nonresponse to treatment with diazoxide. Monoallelic variants result in a variable degree of KATP channel dysfunction, ranging from severe dysfunction associated with a poor response to diazoxide and mild dysfunction associated with a favourable response.
The pattern of inheritance is also associated with the severity of histological abnormalities in the pancreas. Thus, the presence of a single potassium channel variant inherited from the father combined with a de novo somatic mutation in region 11p15 results in the absence of KATP channels in localised regions of the pancreas (focal lesions). Thus, the detection of a single heterozygous variant inherited from the father is a strong predictor of focal lesions. In some case series, this pattern is confirmed in up to 74% of cases.9 In other studies, the presence of a paternally inherited variant predicted the development of focal lesions with a sensitivity of 97% and a specificity of 90%.11
An early aetiological diagnosis is important. While the approach to diagnosis varies between centres, in general panels for detection of changes in the genes that encode potassium channels (ABCC8 and KCNJ11) are used in patients that do not respond to a full course of diazoxide (15–20 mg/kg/day for at least 5 days). Some studies7 have found changes in these genes in up to 88% of cases. This strategy allows early identification of patients with focal lesions, who can then benefit from early and usually curative surgery.7,9
For patients that respond well to treatment with diazoxide, testing is generally done with larger panels that include genes such as ABCC8, KCNJ11, GLUD1, GCK, HADH, UCP2, HNF4α, HNF1α and MCT1.
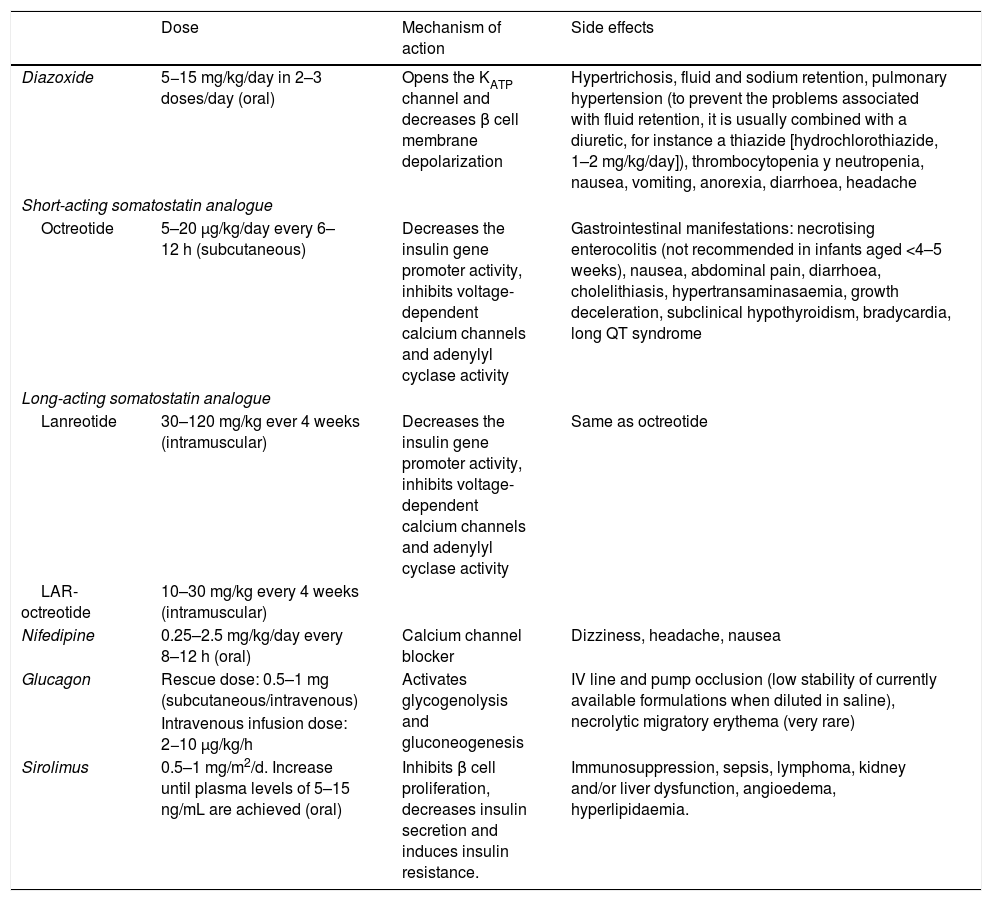
The treatment is complex and delivered by a multidisciplinary team, and includes administration of glucose at high doses, drugs (Table 1) acting on β cell membrane stability and, in some cases, surgery. Families need psychological support and help with the transition to life outside the hospital, which requires collaboration with social workers and psychologists.
Drugs used in the management of congenital hyperinsulinism.
| Dose | Mechanism of action | Side effects | |
|---|---|---|---|
| Diazoxide | 5−15 mg/kg/day in 2–3 doses/day (oral) | Opens the KATP channel and decreases β cell membrane depolarization | Hypertrichosis, fluid and sodium retention, pulmonary hypertension (to prevent the problems associated with fluid retention, it is usually combined with a diuretic, for instance a thiazide [hydrochlorothiazide, 1–2 mg/kg/day]), thrombocytopenia y neutropenia, nausea, vomiting, anorexia, diarrhoea, headache |
| Short-acting somatostatin analogue | |||
| Octreotide | 5–20 μg/kg/day every 6–12 h (subcutaneous) | Decreases the insulin gene promoter activity, inhibits voltage-dependent calcium channels and adenylyl cyclase activity | Gastrointestinal manifestations: necrotising enterocolitis (not recommended in infants aged <4–5 weeks), nausea, abdominal pain, diarrhoea, cholelithiasis, hypertransaminasaemia, growth deceleration, subclinical hypothyroidism, bradycardia, long QT syndrome |
| Long-acting somatostatin analogue | |||
| Lanreotide | 30–120 mg/kg ever 4 weeks (intramuscular) | Decreases the insulin gene promoter activity, inhibits voltage-dependent calcium channels and adenylyl cyclase activity | Same as octreotide |
| LAR-octreotide | 10–30 mg/kg every 4 weeks (intramuscular) | ||
| Nifedipine | 0.25–2.5 mg/kg/day every 8–12 h (oral) | Calcium channel blocker | Dizziness, headache, nausea |
| Glucagon | Rescue dose: 0.5–1 mg (subcutaneous/intravenous) | Activates glycogenolysis and gluconeogenesis | IV line and pump occlusion (low stability of currently available formulations when diluted in saline), necrolytic migratory erythema (very rare) |
| Intravenous infusion dose: 2−10 μg/kg/h | |||
| Sirolimus | 0.5–1 mg/m2/d. Increase until plasma levels of 5–15 ng/mL are achieved (oral) | Inhibits β cell proliferation, decreases insulin secretion and induces insulin resistance. | Immunosuppression, sepsis, lymphoma, kidney and/or liver dysfunction, angioedema, hyperlipidaemia. |
We conducted a retrospective review of patients with a diagnosis of CHI managed in our unit in the past 18 years (2001–2018). Since our hospital is the referral hospital at the regional level, we receive patients with severe disease from the islands of Gran Canaria, Lanzarote and Fuerteventura.
In patient 1, due to clinical suspicion (glucose levels in the diabetic range during adolescence), testing targeted genes HNF1α and HNF4α. In cases 2, 3 and 4, testing targeted the genes involved in the potassium channel (ABCC8 and KCJN11). In all other patients, genetic testing screened for changes in 11 genes (ABCC8, GCK, GLUD1, HADH, HNF1α, HNF4α, INSR, KCNJ11, SLC16A1, UCP2 and SLC25A15). Testing was performed using next generation sequencing (Illumina system) and the results interpreted with a variety of bioinformatic tools based on the clinical presentation of the patient.
During the period under study, according to the Instituto Canario de Estadística (ISTAC) and based on data from the Instituto Nacional de Estadística (INE), there were a total of 130 095 births in the island of Gran Canaria and 26 408 in the island of Lanzarote.
ResultsSince 2001, we have managed 8 cases of CHI with a positive genetic test and 2 in which no variant classified as pathogenic was detected (patients 9 and 10). Of these 10 patients, one resided in the island of Lanzarote and 9 in the island of Gran Canaria. Table 2 presents the clinical characteristics of the patients. Table 3 presents the results of laboratory and genetic testing.
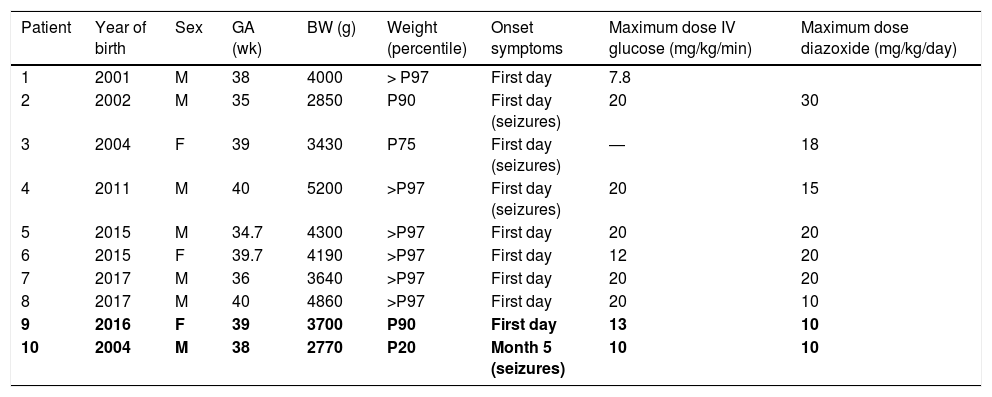
Clinical characteristics.
| Patient | Year of birth | Sex | GA (wk) | BW (g) | Weight (percentile) | Onset symptoms | Maximum dose IV glucose (mg/kg/min) | Maximum dose diazoxide (mg/kg/day) |
|---|---|---|---|---|---|---|---|---|
| 1 | 2001 | M | 38 | 4000 | > P97 | First day | 7.8 | |
| 2 | 2002 | M | 35 | 2850 | P90 | First day (seizures) | 20 | 30 |
| 3 | 2004 | F | 39 | 3430 | P75 | First day (seizures) | ― | 18 |
| 4 | 2011 | M | 40 | 5200 | >P97 | First day (seizures) | 20 | 15 |
| 5 | 2015 | M | 34.7 | 4300 | >P97 | First day | 20 | 20 |
| 6 | 2015 | F | 39.7 | 4190 | >P97 | First day | 12 | 20 |
| 7 | 2017 | M | 36 | 3640 | >P97 | First day | 20 | 20 |
| 8 | 2017 | M | 40 | 4860 | >P97 | First day | 20 | 10 |
| 9 | 2016 | F | 39 | 3700 | P90 | First day | 13 | 10 |
| 10 | 2004 | M | 38 | 2770 | P20 | Month 5 (seizures) | 10 | 10 |
BW, birth weight; F, female; GA, gestational age; M, male; P, percentile.
The data for patients in whom a pathogenic variant was not detected (patients 9 and 10) is presented in boldface.
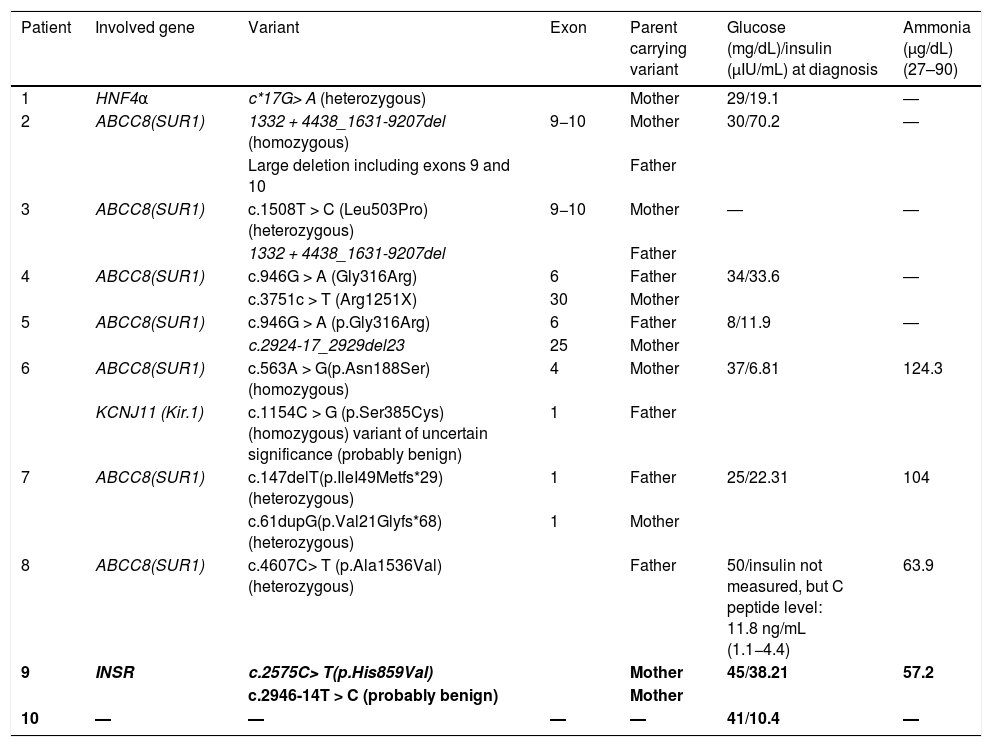
Results of genetic and laboratory tests.
| Patient | Involved gene | Variant | Exon | Parent carrying variant | Glucose (mg/dL)/insulin (μIU/mL) at diagnosis | Ammonia (μg/dL) (27–90) |
|---|---|---|---|---|---|---|
| 1 | HNF4α | c*17G> A (heterozygous) | Mother | 29/19.1 | ― | |
| 2 | ABCC8(SUR1) | 1332 + 4438_1631-9207del (homozygous) | 9−10 | Mother | 30/70.2 | ― |
| Large deletion including exons 9 and 10 | Father | |||||
| 3 | ABCC8(SUR1) | c.1508T > C (Leu503Pro) (heterozygous) | 9−10 | Mother | ― | ― |
| 1332 + 4438_1631-9207del | Father | |||||
| 4 | ABCC8(SUR1) | c.946G > A (Gly316Arg) | 6 | Father | 34/33.6 | ― |
| c.3751c > T (Arg1251X) | 30 | Mother | ||||
| 5 | ABCC8(SUR1) | c.946G > A (p.Gly316Arg) | 6 | Father | 8/11.9 | ― |
| c.2924-17_2929del23 | 25 | Mother | ||||
| 6 | ABCC8(SUR1) | c.563A > G(p.Asn188Ser) (homozygous) | 4 | Mother | 37/6.81 | 124.3 |
| KCNJ11 (Kir.1) | c.1154C > G (p.Ser385Cys) (homozygous) variant of uncertain significance (probably benign) | 1 | Father | |||
| 7 | ABCC8(SUR1) | c.147delT(p.IleI49Metfs*29) (heterozygous) | 1 | Father | 25/22.31 | 104 |
| c.61dupG(p.Val21Glyfs*68) (heterozygous) | 1 | Mother | ||||
| 8 | ABCC8(SUR1) | c.4607C> T (p.Ala1536Val) (heterozygous) | Father | 50/insulin not measured, but C peptide level: 11.8 ng/mL (1.1−4.4) | 63.9 | |
| 9 | INSR | c.2575C> T(p.His859Val) | Mother | 45/38.21 | 57.2 | |
| c.2946-14T > C (probably benign) | Mother | |||||
| 10 | ― | ― | ― | ― | 41/10.4 | ― |
Novel variants presented in italics.
The data for patients in whom a pathogenic variant was not detected (patients 9 and 10) is presented in boldface.
Seventy-five percent (6) of the patients were male. Seven were white (87.5%) and 1 of Indian origin (12.5%).
When it came to the weight at birth, all patients had weights above the 75th percentile and 87% above the 90th percentile (7 cases).
In the past 18 years, the combined incidence of CHI in Gran Canaria and Lanzarote is 1 per 19,518 live births (1/15,614 for the two islands if we include patients in whom genetic testing did not detect pathogenic variants).
The mean level of insulin detected in our sample in the context of hypoglycaemia (glucose ≤45 mg/dL) was 18.7 μIU/mL, and the median was 20.7 μIU/mL (range, 6.8–70.2). We did not find differences between patients in whom testing detected a pathogenic variants and patients in whom it did not.
In the subset of patients in whom testing detected pathogenic variants, 7 had changes in the ABCC8 gene (87%) and 1 in the HNF4α gene (13%). Three patients with a variant in the ABCC8 gene (patients 2, 3 and 5) and the patient with a variant in the HNF4α gene (patient 1) had variants that had not been previously described (Table 3).
All patients with changes in the ABCC8 gene required intravenous glucose at high doses (mean, 16 mg/kg/min; range, 12–20 mg/kg/min). We were not able to classify patients as respondents or nonrespondents to treatment with diazoxide because patients did not undergo the fasting test after reaching the maximum dose. Six patients exhibited a partial response to diazoxide (decreased requirements of IV glucose) and the other 2 did not respond (patients 2 and 7). We only initiated treatment with octreotide in patient 7, who exhibited a partial response. At present (age 4 years) this patient is being treated with intramuscular lanreotide (30 mg/28 days), which has achieved a decrease in the frequency of hypoglycaemic episodes and greater stability in blood glucose levels since treatment initiation.
Patient 1, with a HNF4α variant, had the typical presentation associated with changes in this gene: hypoglycaemia secondary to hyperinsulinism in the neonatal period and blood glucose levels in the diabetic range following the onset of puberty. In the neonatal period, the patient received IV glucose until day 18 post birth at a lower dose than patients with changes in the ABCC8 gene (of up to 7.8 mg/kg/min). The patient needed feedings every 3 hours fortified with maltodextrin through age 2–3 months. This patient did not receive pharmacological treatment. At age 12 years, the patient underwent a glucose tolerance test, with a fasting blood glucose level of 119 mg/dL and a level of 259 mg/dL 2 h after administration of glucose. Repeat blood tests showed similar values later on. The concentration of glycated haemoglobin HbA1c remained below 5.5% at all times and pancreatic autoantibodies were negative (GAD, IA2 and insulin autoantibodies). The variant was inherited from the mother (who was asymptomatic). It was an intronic variant, c*17G > A, located in the 3'-UTR region. The prediction algorithm Mutation Taster assigned this variant a high probability of pathogenicity of 0.92 (Table 4). Functional assays were not performed to confirm pathogenicity.
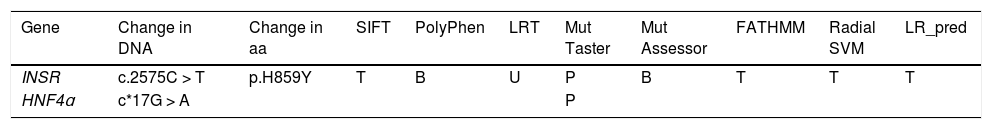
Results of prediction algorithms for the changes detected in the INSR and HNF4ɑ genes (patients 9 and 1, respectively).
| Gene | Change in DNA | Change in aa | SIFT | PolyPhen | LRT | Mut Taster | Mut Assessor | FATHMM | Radial SVM | LR_pred |
|---|---|---|---|---|---|---|---|---|---|---|
| INSR | c.2575C > T | p.H859Y | T | B | U | P | B | T | T | T |
| HNF4ɑ | c*17G > A | P |
aa, amino acid; B, benign variant; P, pathogenic variant; T, tolerated variant; U, variant of uncertain significance.
Description of the changes detected in the INSR y HNF4ɑ genes: affected gene, DNA cytoband, changes in amino acid sequence and computational prediction tools (SIFT, PolyPhen, LRT, Mutation Taster, Mutation Assessor, FATHMM, Radial SVM and LR_pred for INSR and Mutation Taster para HNF4ɑ).
When it came to the degree of pancreatic involvement, our hospital did not have the resources to perform PET-L-DOPA imaging, so patients were referred to national reference centres (La Paz and Vall d’Hebron hospitals) to assess the need for surgery. The PET scan was performed in 4 patients (patients 2, 5, 7 and 8) and found diffuse involvement in all.
Two of the patients (patients 2 and 7) underwent a subtotal pancreatectomy, both with a variant in the ABCC8 and evidence of diffuse involvement in the PET scan. Patient 2 started to exhibit blood glucose levels in the diabetic range a few weeks after the surgery that required administration of insulin, a treatment that persists to date. Patient 7 had a favourable initial response followed by recurrence, and treatment with lanreotide was initiated when the patient was approximately 14 months of age.
Patients in whom no pathogenic variants were identified required a mean dose of IV glucose of 11.5 mg/kg/min (range, 10–13 mg/kg/min). Patient 9 had 2 variants in the INSR gene, both of which were also present in the mother (from South America). The relevant personal history of the patient included meconium aspiration at birth. The mother had never had any symptoms indicative of hypoglycaemia. The c.2946-14T > C variant (inherited, heterozygous and intronic) was classified as benign by prediction algorithms. Variant c.2575 > T (p.H859Y) (inherited, heterozygous and exonic) had not been described previously, has a low allele frequency and was classified as pathogenic by Mutation Taster. It maps to the β subunit of the insulin receptor in a highly conserved region far from the binding site. The gnomAD database lists a frequency of 1:125 in the African population and reports at least 1 case of this variant found in homozygosis in Latin America. When we used other prediction algorithms, we found that SIFT and PolyPhen- 2 classify it as benign and all others as a tolerated variant (Table 4).
Since the mother also carried the variant, we performed an oral glucose tolerance test in the mother that revealed plasma glucose and insulin levels in the normal range (Table 5).
Results of glucose tolerance test in the mother of the patient with a INSR variant.
| +30 min | +60 min | +90 min | +120 min | +180 min | +240 min | +300 min | ||
|---|---|---|---|---|---|---|---|---|
| Glucose (mg/dL) | 89 | 130 | 84 | 97 | 92 | 62 | 75 | 87 |
| Insulin (μU/mL) | 5.11 | 49.12 | 23.22 | 16.53 | 14.2 | 4.59 | 2.99 | 3.35 |
| C peptide (ng/mL) | 2.06 | 7.69 | 5.72 | 5.14 | 5.17 | 3 | 1.95 | 1.75 |
The large number of patients with CHI in our area in recent years motivated us to carry out the review presented in this article and analysis of the cases managed in our hospital.
When we compared our findings with those in the rest of the Spanish territory, we were surprised by the fact that all our patients received the diagnosis within 24 h from birth, due to the presence of symptoms (convulsive seizures) or to the detection of hypoglycaemia in routine tests. Martínez et al.12 reported diagnosis in the first month of life in only 52% of the patients under study. Guerrero-Fernández et al.13 reported that 77% of the patients described in their article had symptoms in the first week of life. Fernández-Marmiesse et al.14 reported diagnosis in every patient in the first 3 days post birth.
Except for patient 3, whose birth weight was in the 75th percentile, the rest of the patients in the case series had birth weights above the 90th percentile (87.5%). Martínez et al.12 reported high birth weights in only 34% of the cases, Guerrero-Fernández et al.13 in 27% and Fernández-Marmiesse et al.14 in 45%.
As for the sex distribution, 75% of the patients in our case series (6/8) were male. Larger samples found lower proportions of male patients, such as those described by Guerrero-Fernández et al. or Martínez et al., of 54% and 64%, respectively.
The incidence of 1 case per 19,518 live births is one of the highest ever reported, higher than the incidence reported in the Netherlands in 1990 (1/50 000)1 or Japan in 2011 (1/35 400)2 and lower than the one reported in 1998 in Saudi Arabia (1/2500).3 Older sources1,3 calculated the incidence based on a clinical diagnosis, without genetic confirmation. If we made a similar calculation, including the 2 patients in whom testing did not detect changes in previously identified genes, the incidence would increase to 1 per 15,614 live births.
In our case series, the gene involved most frequently was ABCC8 (87% of cases), which was similar to the frequency observed by Martinez et al.12 (78%) and somewhat higher than the one reported by Fernández-Marmiesse et al.14 (68%) for the Spanish population. International authors have reported similar proportions. Kapoor et al.9 found variants in the ABCC8 gene in 79% of patients with CHI in whom testing had detected genetic changes. In the largest series published to date, Rosenfeld et al.7 reported that 85% of patients with confirmed genetic changes had variants in the ABCC8 or KCJN11 gene.
Until 2006, the homozygous large deletion 1332 + 4438_1631-9207del affecting exons 9 and 10 of the ABCC8 gene had only been described in one of our patients.14 Later on, it was found in heterozygosis in patient 3, who had inherited it from the father. Patient 2 is from Lanzarote. Patient 3 is from Gran Canaria, but her father and paternal grandparents are from Lanzarote.
The deletion c.2924-17_2929del23 found in patient 5 was also a novel variant. Since most prediction algorithms have been designed to classify missense mutations, we were not able to apply them to these 2 variants. We were also unable to carry out functional assays to confirm their pathogenicity. However, given the location of these changes, which affect the intron-exon junction, they are likely to have a deleterious effect.
When it comes to the findings of genetic testing in patient 9, there have been previous descriptions of changes in the INSR gene causing hypoglycaemic episodes secondary to hyperinsulinism in patients aged up to 3 years, but not in the neonatal period.15 Based on the results of prediction algorithms, the maternal history and the results of the tests performed in the mother, we tend to believe that this is a benign variant unlikely to have a functional impact.
As regards the degree of pancreatic involvement, 4 patients underwent assessment with PET-L-DOPA (patients 2, 5, 7 and 8). Patient 2 had the homozygous large deletion 1332 + 4438_1631-9207del, and patients 5 and 7 had heterozygous variants inherited from the mother and father. Given the pattern of inheritance, the type of pancreatic involvement found in these patients was to be expected. However, in patient 8, who had a heterozygous variant inherited from the father, the findings were unexpected. We had expected a focal lesion pattern based on the findings published by Snider et al.11 In any case, other authors9 have reported diffuse involvement in up to 26% of patients with heterozygous variants inherited from the father.
In conclusion, we ought to underscore the high incidence of CHI in our population. Patients in our hospital received an earlier diagnosis and had greater birth weights compared to other studies conducted in Spain. The gene involved most frequently was ABCC8. Twenty percent of patients required a pancreatectomy. Four had novel variants. Two of them had the large deletion 1332 + 4438_1631-9207del in the ABCC8 gene, and the source of the variant in both patients was identified as the island of Lanzarote (founder effect). All patients had insulin levels of 6.81 μIU/mL or greater at the time of diagnosis.
Conflicts of interestThe authors have no conflicts of interest to declare.
We thank Dr Carrascosa, Dr Gussinyé, Dr Yeste, Dr Clemente and the rest of the paediatric endocrinology team of the Hospital Vall d’Hebron (Barcelona), as well as Dr González Casado, Dr Salamanca and the rest of the paediatric endocrinology team of the Hospital La Paz (Madrid) for their help and guidance in the management of our most complex patients. We also want to thank Dr de León, Dr Stanley and their team at the Philadelphia Children’s Hospital for their support and advice.
Please cite this article as: Nóvoa-Medina Y, Domínguez García A, Quinteiro González S, García Cruz LM, Santana Rodríguez A. Hiperinsulinismo congénito en Gran Canaria. An Pediatr (Barc). 2021;95:93–100.
