



The aetiology of Kawasaki disease (KD) remains unknown. Several studies have linked the human microbiome with some diseases. However, there are limited studies on the role of the respiratory microbiome in KD. The aim of our study was to make a more thorough analysis of the causes and processes that increase the susceptibility to KD.
MethodsCase-control study comparing the respiratory microbiome of KD patients with that of healthy children. The V3–V4 region of the 16S rRNA bacterial gene and 16 respiratory viruses were analysed by real-time polimerase-chain reaction. We used the Ribosomal Database Project (RDP) version 11.5 (taxonomic assignment).
ResultsThe initial sample included 11 cases and 11 controls matched for age, sex and seasonality. One of the cases was excluded to poor sample quality. The final analysis included 10 cases and 10 controls. In the case group, the analysis detected Haemophilus, Moraxella, Streptococcus and Corynebacterium species (27.62%, 19.71%, 25.28%, 11.86%, respectively). In the control group, it found Haemophilus, Streptococcus, Moraxella, and Dolosigranulum species (38.59%, 23.71%, 16.08, 8.93%, respectively). We found a higher relative abundance of Corynebacterium in patients with KD (11.86% vs. 1.55%; P = 0.004).
ConclusionsTo our knowledge, this is the first study that has found differences in the composition of the respiratory microbiome between patients with KD and healthy controls. The relative abundance of Corynebacterium spp. was greater in the KD group. This study shows differences in the microbiome between cases and controls, which suggests that the microbiome may play a role in facilitating the development of KD.
La etiología de la enfermedad de Kawasaki (EK) sigue siendo desconocida. Varios estudios han relacionado el microbioma humano con algunas enfermedades. Sin embargo, los estudios sobre el microbioma respiratorio en EK son limitados. Este estudio intenta profundizar en las causas y procesos que predisponen al desarrollo de la EK.
MétodosEstudio de casos y controles en el que se compara el microbioma respiratorio de pacientes con EK con el de niños sanos. La región V3-V4 del gen bacteriano del ARNr 16S y 16 virus respiratorios se analizaron mediante reacción en cadena de la polimerasa en tiempo real. Se utilizó la base de datos RDP (Ribosomal Database Project) versión 11.5 (asignación taxonómica).
ResultadosSe incluyeron 11 casos y 11 controles emparejados por edad, sexo y estacionalidad. Uno de los casos fue descartado por mala calidad de la muestra. El estudio final se realizó a 10 casos y 10 controles. En el grupo de casos se encontraron Haemophilus, Moraxella, Streptococcus y Corynebacterium (27,62%, 19,71%, 25,28% y 11,86%, respectivamente). En el grupo control, Haemophilus, Streptococcus, Moraxella y Dolosigranulum (38,59%, 23,71%, 16,08 y 8,93%, respectivamente). Corynebacterium mostró una mayor abundancia en pacientes con EK (11,86% vs. 1,55%; p = 0,004).
ConclusionesHasta donde sabemos, este es el primer estudio que ha encontrado diferencias en la composición del microbioma respiratorio entre pacientes con EK y controles sanos. Corynebacterium spp. presentó una mayor abundancia en el grupo de EK. Este estudio muestra diferencias en el microbioma entre pacientes y controles, lo que sugiere un papel facilitador del microbioma en el desarrollo de la EK.
Kawasaki disease (KD) is an acute self-limited systemic vasculitis of childhood of unknown aetiology that presents predominantly in infants and children under 5 years old. Its diagnosis is based on clinical criteria. Its prognosis depends on the extent of cardiac involvement. Without treatment, up to 20%–25% of patients develop coronary aneurysms.1
The aetiology of KD is unknown despite years of investigation. Clinical, laboratory and epidemiological features suggest an infectious origin or trigger. To date, many studies have failed to identify an infectious aetiological agent or demonstrate an association with exposure to drugs or a response to super-antigen. Besides, there is evidence of an important genetic predisposition for KD. Some genome-wide association studies (GWAS) have identified some biologically plausible loci involved in inflammation, immune responses and cardiovascular status.2–4 Due to the clustering of cases in space and/or time, among other patterns, a reasonable hypothesis is that KD may be caused by a trigger (whether it is an infectious agent is still under debate) that causes the disease or elicits a response in genetically predisposed individuals, particularly those of Asian descent.1 However, little is known about the impact of the human microbiome in the development of KD.
The term microbiome refers to the community of commensal, symbiotic and pathogenic microorganisms that share human body space.5 Several studies have linked the human microbiome with some diseases or the predisposition to develop them. Some studies have aimed at establishing an association between the dysregulation of the gut microbiome and multiple autoimmune diseases. Animal models suggest that the gut microbiome contains organisms that direct both pro-inflammatory and anti-inflammatory immune responses depending on the immunologic context, genetic makeup or sex of the host and the overall microbial community structure.6
The role of human microbiome in rheumatology has been studied in diseases such as rheumatoid arthritis (RA) or systemic lupus erythematosus (SLE). In the case of RA, evidence has been found of an association between the increased presence of Porphyromonas gingivalis, an oral anaerobe involved in the development of periodontitis, and positive anticitrullinated peptide antibodies.7 Gut microbial perturbations are present in RA too, with substantial expansion of Prevotella copri in untreated patients with new-onset RA.8 Systemic lupus erythematosus is also characterized by microbial perturbations. One of the earliest studies found decreased gut bacterial diversity and a lower Firmicutes to Bacteroidetes ratio in patients with SLE.9 As occurs in RA, there is evidence of translocation of intestinal bacteria in SLE, with detection of Enterococcus gallinarum in liver biopsies of patients with active SLE.10
Although the involvement of dysregulation of the intestinal microbiome in the pathogenesis of autoimmune diseases has been widely studied, the role of the respiratory microbiome in autoimmunity is not as well established. Although no infectious causative agent has been identified, some epidemiological studies have documented infections by various microorganisms in many KD patients. Recently, KD-like cases have been described in association with SARS-CoV-2 infection.11,12 Besides, the fact that there have been KD epidemics in Japan, its higher incidence in spring and winter and its occurrence predominantly in children aged 6 months to 5 years, support the hypothesis of an infectious aetiology or trigger.1
To our knowledge, studies on the role of the microbiome in KD are limited. Some studies sought to establish a link between the intestinal and pharyngeal flora with KD and found no significant differences between cases and controls.13,14 In a study on the intestinal microbiome in KD published in 2005, the faecal microbiome of 28 KD patients was analysed, evincing an increase of Streptoccocus spp. in the acute phase of the disease and Ruminococcus bacteria in the subacute phase.15 Recent advances allowed the discovery that Th17 and Treg cell differentiation is regulated by short chain fatty acids (SCFAs), in particular butyrate, produced by the gut microbiota. This finding provided a mechanistic link between dysbiosis, defined as changes in the composition of the gut microbiota, and various inflammatory diseases. Recent studies have tried to support this hypothesis, finding that faecal concentrations of butyrate were significantly reduced in KD patients, providing important information regarding the association between dysbiosis and dysregulated immune responses in KD.15–17
The contribution of viruses to the pathogenesis of KD has been suggested by ultrastructural studies that found cytoplasmic inclusion bodies containing RNA of viral origin in the bronchial epithelia of deceased KD patients.18–20 Other studies have found that one or more respiratory viruses were detected in nasopharyngeal aspirates of approximately half of KD patients.21
Some works have reported a possible role of fungal infections in the development of KD. Recent research22,23 has suggested that the agent causing KD may be an environmental agent carried through tropospheric winds. The authors found aerosolized toxins from several Candida species in specimens collected in the troposphere at times of high KD incidence. In Japan, the burden of cases often appears to be associated with wind currents originating in densely cultivated cereal croplands of north-eastern China.24,25 In addition, Sato et al. observed that Candida albicans-derived substances, such as C. albicans water-soluble fraction, induced coronary arteritis in mice.26
Most of the data shows that the composition of the gut microbiome in KD patients differs from that in healthy subjects.16 A plausible hypothesis for the pathogenesis of KD would be that, in genetically predisposed individuals with a favourable microbiome environment, an external trigger could facilitate the development of the disease. As far as we know, there are no studies on the characterization of the microbiome found in respiratory exudates in KD.
The main objective of this study was to compare differences in the nasopharyngeal bacterial microbiota and respiratory viruses between KD patients and healthy controls as a first step to delve deeper into the role of the microbiome and previous infections in the pathogenesis of KD.
Material and methodsWe conducted a case-control study including children (cases) diagnosed with KD treated in a tertiary care hospital (n = 11) matched with healthy children (controls) managed in the same hospital and who underwent routine laboratory tests previous to minor surgery or general paediatrics follow-up visits (n = 11). One of the cases was eventually excluded due to poor sample quality. The final study was performed in 10 cases and 10 controls. Samples were collected between February 2016 and May 2018. Cases and controls were matched for age, sex and seasonality. Additional information on the methods can be found in the Supplementary material.
All patients were enrolled in the 5 days that followed the diagnosis, and the respiratory samples were collected before initiation of treatment. They all meet the diagnostic criteria for KD established by the American Heart Association.1 We excluded patients that had received antibiotics before sample collection or without a completed informed consent form. Controls were healthy children without major comorbidities and who had not received antibiotic treatment in the 4 weeks preceding sample collection. The data sets presented in this article can be found in online repositories. The names of the repository/repositories and accession number(s) are detailed in the Figshare repository within the “Kawasaki” project: https://figshare.com/s/181b728556419d0de4c5.
Extraction of nasopharyngeal aspirate samplesA nasopharyngeal aspirate sample was collected from each patient for testing. To do so, 2–3 mL of Meinsol® 9 mg/mL sodium chloride (Fresenius Kabi; Germany) were injected and aspirated from the patient’s nasopharynx and collected in 25 mL sterile tubes. Samples were aliquoted into 1.5 mL Eppendorf tubes (STARLAB International GmbH, Hamburg, Germany) and immediately frozen at –80 °C. Six negative control samples (2−3 mL of Meinsol® 9 mg/mL sodium chloride collected in sterile tubes) were also processed. Both clinical samples and negative controls were subjected to the same protocol of collection, genomic DNA extraction, amplification and sequencing. Sample processing was carried out in a clinical diagnostic laboratory with nucleic acid-free areas and under UV-treated laminar flow hoods, with reagents and pipettes allocated exclusively to this process.
DNA/RNA extractionDNA extraction from aspirates and negative controls was performed using Nuclisens® EasyMag® following the directions of the manufacturer (bioMérieux; Marcy-l’Étoile, France). This is an automated nucleic acid extraction system based on magnetic silica particles. From a sample volume of 400 μL, a final elution volume of 25 μL is obtained. The amount of DNA extracted was subsequently quantified using Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific; Massachusetts, USA), a fluorometric quantification method based on the specific binding of fluorochromes to double-stranded DNA. The extraction of DNA/RNA for respiratory virus detection was performed with Magna Pure (Roche Diagnostics; Indianapolis, IN, USA), another automated extraction system also based on magnetic silica particles, according to the manufacturer’s instructions.
Study of the microbiome by next-generation sequencing (NGS)The V3–V4 region of the 16S ribosomal RNA gene (16S rRNA), of approximately 465 base pairs, was amplified by polymerase chain reaction (PCR) using specific primers and with the following cycling conditions: 95 °C (3 min), followed by 25 cycles at 95 °C (30 s), 55 °C (30 s), 72 °C (30 s) and 72 °C (5 min). Afterwards, we proceeded to the fragmentation of the total DNA of each sample and the addition of the two indexes and sequencing adapters with Nextera XT Illumina Index Kit (Illumina, San Diego, California, USA) using PCR. A second purification was carried out with Agencourt AMPure XP 60 mL kit beads (Beckman Coulter; Munich, Germany) prior to quantification of the library with the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific; Massachusetts, USA). A library of 300 bp clones was constructed for each sample. An equimolar amount of DNA from each sample was pooled and sequenced with the Illumina MiSeq system (Illumina; San Diego, California, USA).
Detection of respiratory virusesWe used a multiplex real-time (RT) PCR assay (Allplex Respiratory Panel) to detect 16 respiratory viruses and 3 subtypes of influenza A virus that cause respiratory tract infections: influenza A (H1, H1pdm09 and H3 subtypes), influenza B, respiratory syncytial virus A and B, adenovirus, enterovirus, parainfluenza virus 1, 2, 3 and 4, metapneumovirus, bocavirus, rhinovirus, coronavirus NL63, coronavirus 229E and coronavirus OC43.
Statistical analysisWe assessed the normality of the data with the Shapiro–Wilk test. We described normally distributed quantitative data as mean and standard deviation (SD). Groups were compared with the Student t test in the case of a normal distribution or the Mann–Whitney U test otherwise. We used Kruskal–Wallis test was to compare more than two groups. We used the Fisher exact test for hypothesis testing with qualitative variables. Statistical significance was defined as a P-value of less than 0.05.
Ethical considerationsThe study was approved by the Ethics Committee of our hospital. Every procedure was carried out in compliance with pertinent guidelines and regulations and informed consent was obtained from all participants and/or their legal guardians. The study adhered to the principles of the Declaration of Helsinki.
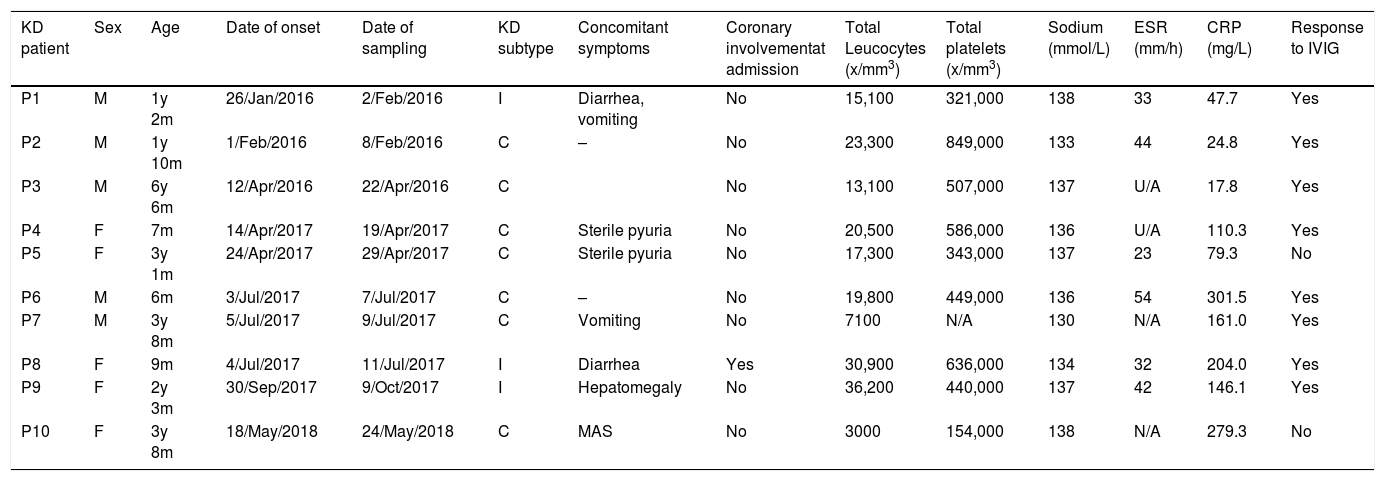
ResultsPatient characteristicsThe study initially included 11 patients with KD (6 males and 5 female), but one was excluded due to poor sample quality; the patients were aged 6–81 months (median, 30 months). All the cases met the diagnostic criteria for KD established by the American Heart Association.1 All patients with KD received intravenous immunoglobulin (IVIG) at a dose of 2 g/kg as first-line treatment, and only 2 required additional treatments, such a second dose of IVIG or corticosteroids. Table 1 presents the clinical characteristics and treatment of the patients. We matched cases and controls for age (same year of birth), sex and seasonality (within the same season). Controls were healthy children without comorbidities that visited the same hospital for follow-up in general paediatrics or prior to minor surgery.
KD patient features at admission. M: male, F: female, y: years, m: months, C: complete, I: incomplete, MAS: macrophage activation syndrome, U/A: unavailable.
| KD patient | Sex | Age | Date of onset | Date of sampling | KD subtype | Concomitant symptoms | Coronary involvementat admission | Total Leucocytes (x/mm3) | Total platelets (x/mm3) | Sodium (mmol/L) | ESR (mm/h) | CRP (mg/L) | Response to IVIG |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | M | 1y 2m | 26/Jan/2016 | 2/Feb/2016 | I | Diarrhea, vomiting | No | 15,100 | 321,000 | 138 | 33 | 47.7 | Yes |
| P2 | M | 1y 10m | 1/Feb/2016 | 8/Feb/2016 | C | – | No | 23,300 | 849,000 | 133 | 44 | 24.8 | Yes |
| P3 | M | 6y 6m | 12/Apr/2016 | 22/Apr/2016 | C | No | 13,100 | 507,000 | 137 | U/A | 17.8 | Yes | |
| P4 | F | 7m | 14/Apr/2017 | 19/Apr/2017 | C | Sterile pyuria | No | 20,500 | 586,000 | 136 | U/A | 110.3 | Yes |
| P5 | F | 3y 1m | 24/Apr/2017 | 29/Apr/2017 | C | Sterile pyuria | No | 17,300 | 343,000 | 137 | 23 | 79.3 | No |
| P6 | M | 6m | 3/Jul/2017 | 7/Jul/2017 | C | – | No | 19,800 | 449,000 | 136 | 54 | 301.5 | Yes |
| P7 | M | 3y 8m | 5/Jul/2017 | 9/Jul/2017 | C | Vomiting | No | 7100 | N/A | 130 | N/A | 161.0 | Yes |
| P8 | F | 9m | 4/Jul/2017 | 11/Jul/2017 | I | Diarrhea | Yes | 30,900 | 636,000 | 134 | 32 | 204.0 | Yes |
| P9 | F | 2y 3m | 30/Sep/2017 | 9/Oct/2017 | I | Hepatomegaly | No | 36,200 | 440,000 | 137 | 42 | 146.1 | Yes |
| P10 | F | 3y 8m | 18/May/2018 | 24/May/2018 | C | MAS | No | 3000 | 154,000 | 138 | N/A | 279.3 | No |
Alpha diversity focuses on the study of diversity within a single sample (intrasample). There are different parameters to measure it, each of which focuses on explaining one or more of its components. In the analysis of α-diversity, we can differentiate between richness and diversity. Richness only takes into account the number of different genera contained in each of the samples, while diversity also takes into account the abundance of these genera in the samples. We analysed different α-diversity indices in both cases and controls, which we have represented as box plots (Fig. 1). We did not find significant differences in either richness or diversity.

Alpha diversity: box plot of the different indices of diversity and richness in the case and control groups. The horizontal line in each plot indicates the median of the data set, and the edges of the box mark the 25% and 75% percentiles. The P-value calculated with the Wilcoxon test is given at the top.
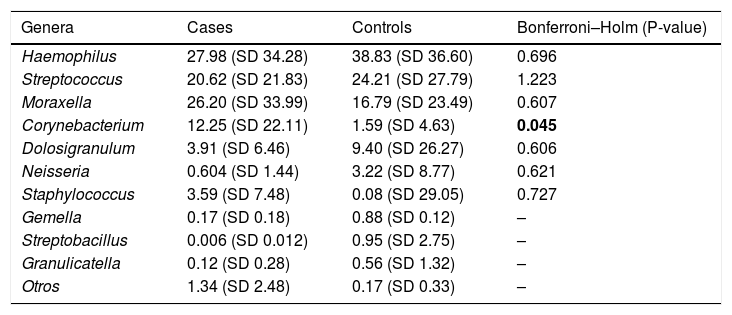
The most frequent genera in the case group were Haemophilus, Streptococcus, Moraxella and Corynebacterium, with a relative abundance of 27.55%, 20.62%, 26.20% and 12.25%, respectively. In comparison, the dominant genus in the control group was Haemophilus, followed by Streptococcus, Moraxella and Dolosigranulum (38.83%, 24.21%, 16.79% and 9.40%, respectively). The rest of the identified genera had relative abundances of less than 4% (Fig. 2 and Table 2).
 abundance of the most frequently detected bacterial genera in both groups. Those bacterial genera with an average relative abundance of less than 0.5% were included in the “other genera” category in the graph.")
Taxonomic bacterial composition of nasopharyngeal samples in the case and control groups: cumulative bar graph showing the relative (average) abundance of the most frequently detected bacterial genera in both groups. Those bacterial genera with an average relative abundance of less than 0.5% were included in the “other genera” category in the graph.
Relative abundances of the genera most frequently detected in cases and controls (mean (SD)). SD: standard deviation.
| Genera | Cases | Controls | Bonferroni–Holm (P-value) |
|---|---|---|---|
| Haemophilus | 27.98 (SD 34.28) | 38.83 (SD 36.60) | 0.696 |
| Streptococcus | 20.62 (SD 21.83) | 24.21 (SD 27.79) | 1.223 |
| Moraxella | 26.20 (SD 33.99) | 16.79 (SD 23.49) | 0.607 |
| Corynebacterium | 12.25 (SD 22.11) | 1.59 (SD 4.63) | 0.045 |
| Dolosigranulum | 3.91 (SD 6.46) | 9.40 (SD 26.27) | 0.606 |
| Neisseria | 0.604 (SD 1.44) | 3.22 (SD 8.77) | 0.621 |
| Staphylococcus | 3.59 (SD 7.48) | 0.08 (SD 29.05) | 0.727 |
| Gemella | 0.17 (SD 0.18) | 0.88 (SD 0.12) | – |
| Streptobacillus | 0.006 (SD 0.012) | 0.95 (SD 2.75) | – |
| Granulicatella | 0.12 (SD 0.28) | 0.56 (SD 1.32) | – |
| Otros | 1.34 (SD 2.48) | 0.17 (SD 0.33) | – |
We found that Corynebacterium spp. were overrepresented in the KD group (12.25% vs 1.59%; P = 0.045). Moreover, a higher proportion of other genera with relative abundances of less than 0.5% was also identified in the case group compared to the control group (16.07% vs 7.82%; P = .08).
Analysis of β-diversityBeta diversity is defined as the difference in species diversity between communities (intersample) and can reveal aspects of microbial ecology that cannot be noticed by simply considering the individual composition of the samples. For β-diversity, we converted operational taxonomic units (OTUs) to relative abundance values, and excluded OTUs corresponding to a relative abundance of less than 0.1%. We did not find significant differences in any of the distance matrices used (P = 0.61) (Fig. 3).
 are groups (clusters) of sequence variants of the 16S rRNA gene. Each cluster is supposed to represent a taxonomic unit of a bacterial species, genus, phylum, or taxa. Depending on the identity threshold between the sequences of a cluster, it can be assumed that the OTU corresponds to a specific species or specific genus. OTUs with 97% similarity correspond to bacterial genera. The OTUs were classified into different taxonomic ranges (domain, phylum, order, family and genus), with a minimum confidence threshold of 80%. The OTU values were converted to relative abundances and OTUs with a relative abundance of less than 0.1% were excluded. We did not find significant differences in any of the distance matrices used (P = 0.61).")
Beta diversity: operational taxonomic units (OTUs) are groups (clusters) of sequence variants of the 16S rRNA gene. Each cluster is supposed to represent a taxonomic unit of a bacterial species, genus, phylum, or taxa. Depending on the identity threshold between the sequences of a cluster, it can be assumed that the OTU corresponds to a specific species or specific genus. OTUs with 97% similarity correspond to bacterial genera. The OTUs were classified into different taxonomic ranges (domain, phylum, order, family and genus), with a minimum confidence threshold of 80%. The OTU values were converted to relative abundances and OTUs with a relative abundance of less than 0.1% were excluded. We did not find significant differences in any of the distance matrices used (P = 0.61).
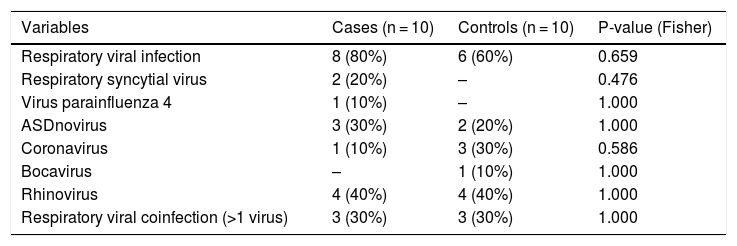
In 80% of the cases of KD (n = 8), the RT-PCR test detected the presence of DNA/RNA of some of the respiratory viruses included in the panel, compared to 60% of the healthy controls (P = 0.659). The most frequently detected viruses in the KD group were rhinovirus (n = 4; 40%), adenovirus (n = 3; 30%), respiratory syncytial virus (n = 2; 20%) and parainfluenza virus 4 (n = 1; 10%), while in the control group, they rhinovirus (n = 4; 40%), coronavirus (n = 3; 30%), adenovirus (n = 2; 20%) and bocavirus (n = 1; 10%). There were no significant differences between groups (Table 3).
Frequency of respiratory viral infection: frequency of respiratory viral infection and individual distribution of viruses analysed by multiple RT-PCR in the case and control groups.
| Variables | Cases (n = 10) | Controls (n = 10) | P-value (Fisher) |
|---|---|---|---|
| Respiratory viral infection | 8 (80%) | 6 (60%) | 0.659 |
| Respiratory syncytial virus | 2 (20%) | – | 0.476 |
| Virus parainfluenza 4 | 1 (10%) | – | 1.000 |
| ASDnovirus | 3 (30%) | 2 (20%) | 1.000 |
| Coronavirus | 1 (10%) | 3 (30%) | 0.586 |
| Bocavirus | – | 1 (10%) | 1.000 |
| Rhinovirus | 4 (40%) | 4 (40%) | 1.000 |
| Respiratory viral coinfection (>1 virus) | 3 (30%) | 3 (30%) | 1.000 |
To our knowledge, this is the first study that has found differences in the composition of the respiratory microbiome between KD patients and healthy controls. In 2004, Horita et al. obtained throat swab samples of 21 patients with KD and 20 controls with non-KD febrile illness. Sixty-one bacterial strains were isolated from KD patients, and 62 strains from control patients. The authors did not find any differences between KD patients and febrile controls in the bacterial species present in their throat flora.13
In our study, we obtained nasopharyngeal aspirate samples of 10 patients with KD and 10 healthy controls and studied the microbiome using NGS. We found a significant difference in the abundance of Corynebacterium, which was overrepresented in the KD group (12.25% vs 1.59%; P = 0.045). Moreover, the proportion of other genera with abundances of less than 0.5% was also higher in the case group compared to the control group (16.07% vs 7.82%; P = 0.08). A previous study that tried to isolate the causative bacterial gene from peripheral blood leukocytes of patients with KD showed a new species of the genus Corynebacterium.27Corynebacterium spp. were also present in aerosol samples collected at ground level in a location very close to Tokyo at a time of high KD incidence. Interestingly, an important number of other taxa previously associated to KD in different studies have also been identified in the metagenomic analysis of airborne samples. This might indicate that potential exposure to air with different loads of bacteria might have a role in the pathogenesis of KD, a subject that deserves further investigation.24,25,28
The genus Corynebacterium is a taxon of coryneform gram-positive bacilli or coccobacilli.29,30 Nearly all are catalase-positive, and they express a wide range of pigments and metabolic processes. Bacteria in this genus are ubiquitous and commonly present in the skin and mucosa of human beings, and most of them are saprophytes. Corynebacterium spp. have been identified in the middle ear of children with otitis media, can cause opportunistic infections in some cases, and a few species in the group, such as C. diphtheriae, are pathogenic for humans.31
Other studies of the gut microbiome have found that the beneficial Lactobacillus genus may be absent or lost from enteric flora during the acute phase of KD14 and that there seems to be a relative increase in Streptococcus spp. in the acute phase of the disease and in Ruminococcus spp. in the subacute phase.15 In our study, we found no differences in any of these 3 genera in the specimens, which could be related to the small sample size or to the fact that we studied respiratory samples rather than faecal samples.
The contribution of viruses to KD has been suggested by studies that found cytoplasmic inclusion bodies containing viral RNA in the bronchial epithelia of deceased KD patients.19 Although several studies have attempted to identify the origin of this viral RNA, no known viruses have been identified, and Rowley et al. concluded that it could be a previously unidentified, ubiquitous RNA virus that can produce persistent infection in the bronchial epithelium, thus contributing to the aetiology of KD.18,20 In another study, Turnier et al. found that nearly half (n = 93, 41.9%) of the 192 patients with KD under study had a positive respiratory viral PCR, mainly rhinovirus or enterovirus.21 Similarly, our study did not found differences between cases and controls, although rhinovirus and adenovirus, among others, where found in nearly half of the samples in both groups.
While there are obvious limitations to our study, mainly the small sample and the lack of comparison with not only healthy controls but also children with respiratory diseases, the results are intriguing if a unique aetiology for this disease is contemplated.
It is possible, though, that KD is a multifaceted disease, and that exposition to a diversity of microbial agents or a dominance of microbial agents with structural similarities might elicit the same idiosyncratic immune response. There is reasonable proof that an imbalance in the microbiome has a role in the pathogenesis of KD. Our study introduces the genus Corynebacterium as a new player in the game and demonstrates the need to conduct larger epidemiological studies focused on uncovering changes in the microbiome of patients with KD.
The current article does not assure causality but establishes the bases for conducting studies with larger samples that help us unravel the role of the Corynebacterium genus in the pathogenesis of KD. Our group is already working on collecting more samples from patients with KD, healthy controls and patients with respiratory infections to try to draw more established conclusions.
ConclusionTo our knowledge, this is the first study that has found differences in the composition of the respiratory microbiome between KD patients and healthy controls. The genus Corynebacterium was significantly more abundant in KD patients compared to healthy controls. These results support the hypothesis that there could be a favourable respiratory microbiome substrate for the development of the disease in genetically predisposed patients. Studies in larger samples are necessary to assess its significance and possible relationship with KD.
FundingThis work was supported by the National R+D+I Plan, Instituto de Salud Carlos III (project PI20/00517) and cofunded by European Regional Development Fund (ERDF). It was also partially funded by a 2018 Sociedad Española de Reumatología Pediatrica (SERPE) scholarship and a 2016 grant from the Fundació Daniel Bravo Andreu to the WINDBIOME project. These funding bodies had no role in study design, data collection and analysis, the decision to publish or the preparation of the manuscript.
Conflicts of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could constitute a conflict of interest.
We are grateful for the support received from the Spanish Ministry of Science and Innovation through the Centro de Excelencia Severo Ochoa 2019–2023 programme (CEX2018 000806S) and from the Government of Catalonia through the CERCA programme.
This work was presented at the XIII Congress of the Spanish Society of Pediatric Rheumatology (SERPE) (Madrid, Spain; November 2019) and the Euro-KiDs 2021 Congress (online meeting; January 2021).
