


In the paediatric age group, hyperkalaemia tends to be asymptomatic, so serum potassium levels greater than 5.5 mEq/L should be verified and investigated, as they may be indicative of potentially severe diseases, as occurred in the case presented here.1
The patient was a boy aged 5 years with a personal history of obesity and type 1 diabetes in whom a blood chemistry panel ordered during a check up revealed a serum potassium level of 6.8 mEq/L in absence of other electrolyte abnormalities. A second test confirmed the finding of hyperkalaemia. Previous blood tests have not found abnormal levels of this ion, and the patient had been asymptomatic at all times.
The assessment was completed with measurement of the arterial blood pressure, which yielded values in the normal range for age and sex. The patient underwent an electrocardiogram that did not evince any abnormalities, as well as venous blood gas analysis that revealed mild metabolic acidosis with a pH of 7.29, a carbon dioxide pressure (pCO2) of 39.3 mmHg, a serum bicarbonate level of 18.2 mEq/L and an excess base of –2.3 mmol/L. We measured basal levels of aldosterone, renin, cortisol and adrenocorticotropic hormone (ACTH), which were normal. We expanded the work up with analysis of a urine sample to calculate the transtubular potassium gradient, which was of 1.8 (normal range, 4–7), which suggested deficient renal secretion of this ion with preserved renal function and a high calcium/creatinine ratio of 0.4 mg/mg (normal range, <0.2 mg/mg).
Retaking the family history revealed that the father had a history of long-term high blood pressure, muscle weakness, calcium renal lithiasis and episodes of atrial fibrillation in the past decade. Both the father and the paternal grandfather (who had died of cancer) had elevated potassium levels in multiple blood tests.
Since the family history and tests performed to date suggested a hereditary aetiology of hyperkalaemia, we approached the illness as a possible case of pseudohypoaldosteronism type II and ordered genetic testing for this syndrome, which confirmed the suspected diagnosis with the detection of a heterozygous change in the KLHL3 gene in both the patient and his father.
Following the diagnosis of pseudohypoaldosteronism type II, despite remaining completely asymptomatic, the patient started treatment with oral hydrochlorothiazide at a dose of 1 mg/kg/day, which achieved normalization of serum levels of potassium and renal calcium excretion, with a favourable outcome thereafter.
In conclusion, if a paediatric patient has elevated potassium levels in 2 or more consecutive blood tests without an apparent cause, the clinician should initiate an aetiological diagnosis.
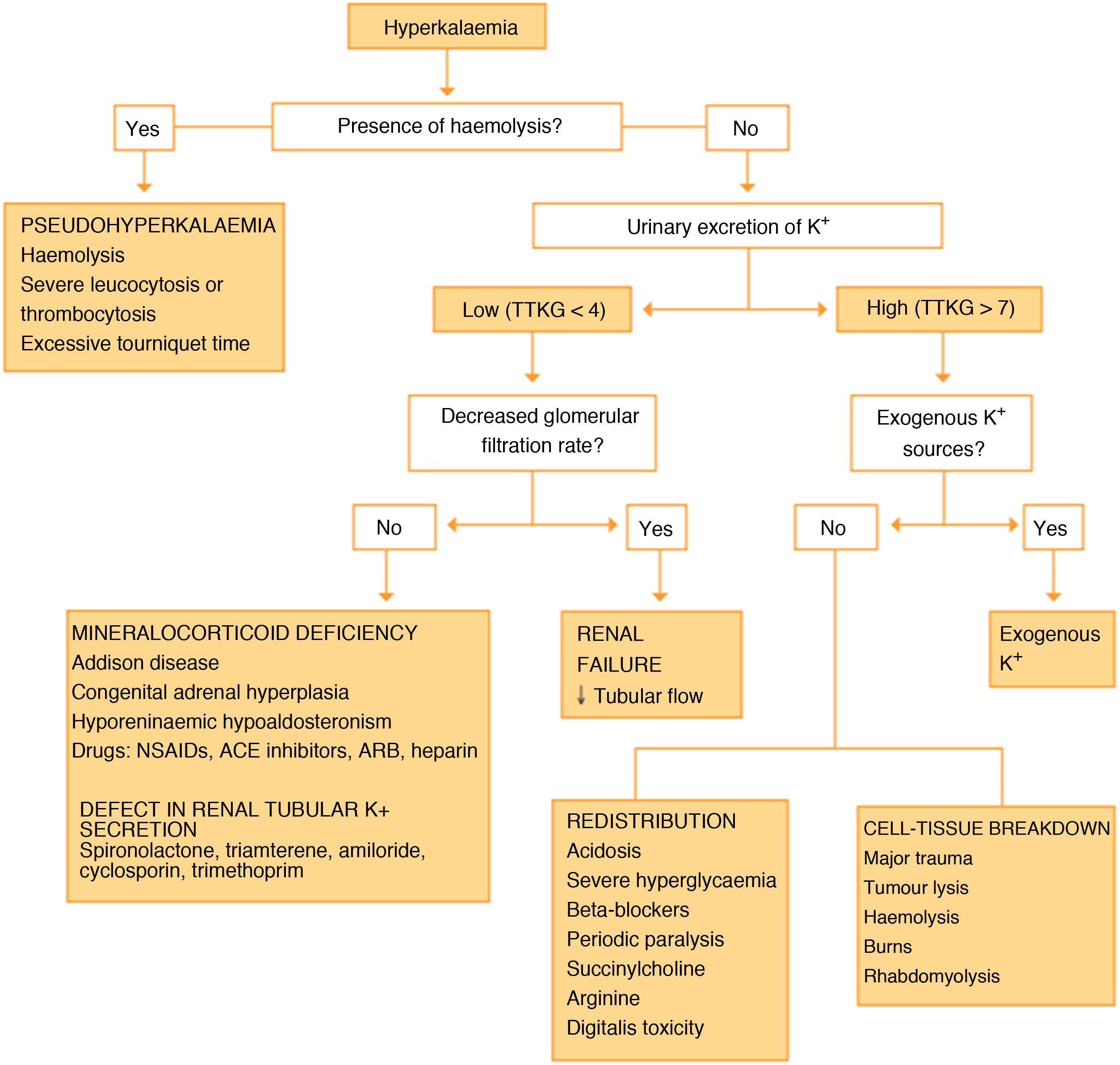
As Fig. 1 shows, to determine the aetiology of the hyperkalaemia, that the work up of these patients should include a urine test to calculate the transtubular potassium gradient and blood tests to measure the glomerular filtration rate and levels of renin, aldosterone, cortisol and ACTH.2 If the transtubular potassium gradient is low (<4) and the glomerular filtration rate is normal, the decreased excretion of this ion by the kidney suggests a mineralocorticoid deficiency or resistance,2,3 and therefore, the presence of normal or slightly elevated levels of renin and aldosterone guides the diagnosis toward unresponsiveness or resistance to the action of aldosterone in target cells.4
Pseudohypoaldosteronism type II, also known as Gordon syndrome, is a rare disease characterised by hyperkalaemia, hypercalciuria and high blood pressure with preserved renal function and normal or slightly elevated levels of aldosterone and renin.2,3 More than 180 families affected by this disease have been reported to date, with a mean age at diagnosis of 26 ± 14 years.2 It is produced by changes in the WNK1, WNK4, CUL3 or KLHL3 gene that affect the renal excretion of sodium and potassium.3,4 In most cases, the pattern of inheritance is autosomal dominant, although cases of autosomal recessive changes in the KLHL3 gene have also been described.3–5 Thiazide diuretics are the first-line treatment and usually achieve adequate control of blood pressure, decreasing the risk of renal lithiasis by decreasing renal excretion of calcium.6
Thus, early diagnosis of pseudohypoaldosteronism type II allows early initiation of treatment, which may modify the long-term outcomes of disease.
Please cite this article as: Pueyo-Agudo E, Cobreros-Pérez Á, Martínez-Rivera V, Nieto-Vega FA, González-Gómez JM, Leiva-Gea I. Hiperpotasemia asintomática como forma de presentación de pseudohipoaldosteronismo. An Pediatr (Barc). 2022;96:263–264.
Previous presentations: The study was presented at the XLIII Congress of the Asociación Española de Nefrología Pediátrica, held May 16–19, 2018 in Valencia, Spain, and the XLVI Scientific Meeting of the Sociedad de Pediatría de Andalucía Oriental, held March 15–16, 2019 in Jaen, Spain.

Anales de Pediatría (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals