




El síndrome tricorrinofalángico (TRPS) es una entidad poco frecuente de herencia autosómica dominante, alta penetrancia y expresividad variable, que se debe a una alteración en el gen TRPS1. Clínicamente se caracteriza por alteraciones del pelo (cabello escaso, adelgazamiento lateral de las cejas) y uñas (distrofia ungueal), leve dismorfia facial (punta nasal bulbosa, filtrum largo y plano, labio superior fino y pabellones auriculares prominentes) y anomalías esqueléticas (talla baja, braquidactilia, desviación de las falanges, epífisis de las falanges en forma de cono, displasia de caderas y osteopenia). Se subdivide en dos tipos: TRPS I (OMIM # 190350), que se debe a variantes patogénicas del gen TRPS1; y TRPS II (OMIM # 150230), que se produce por deleción de genes contiguos en el cromosoma 8 (incluyendo TRPS1 y EXT1) y asocia además osteocondromas y discapacidad intelectual1–3.
En este trabajo presentamos varios miembros de una familia, diagnosticados de TRPS a partir del estudio de un niño de 6 años por hipocrecimiento y ciertos rasgos dismórficos. En el caso índice, a la exploración destacaba una talla de 108,8cm (−2,68 DE), un índice de masa corporal (IMC) de 13,52kg/m2 (p10), con proporciones corporales normales y velocidad de crecimiento de 5,1cm/año. Presentaba cabello claro, fino y escaso, adelgazamiento de la cola de las cejas y uñas frágiles. Además, destacaba la forma triangular de la cara, abombamiento frontal, filtrum largo y plano, labio superior fino y pabellones auriculares grandes y retrovertidos. En las extremidades se observaba clinodactilia del quinto dedo en manos y pies y pulgares de implantación proximal. Además, presentaba hiperlaxitud, con pie plano valgo flexible, y actitud escoliótica sin alteraciones rotacionales vertebrales. Se trataba de un niño nacido a término, con peso al nacimiento de 2690g (p7) y longitud de 49cm (p28), con antecedentes personales de retraso global del desarrollo leve y adenoidectomía por hipertrofia adenoidea.
En la familia no había historia de consanguinidad. Como antecedentes familiares destacaba la talla baja materna 147,2cm (p<1, −2,83 DE) y un fenotipo similar a su hijo. La talla paterna era normal (175cm; p36). Al interrogar a la madre, esta refería que en la familia materna había varios miembros con talla baja y rasgos faciales similares.
El estudio hormonal (IGF1, IGFBP3 y hormonas tiroideas) fue normal y los marcadores de celiaquía negativos. Se observó una edad ósea atrasada 3 años y varias epífisis de las falanges en forma de cono (fig. 1). Se realizó un estudio mutacional de los genes incluidos en panel de displasias óseas, mediante secuenciación masiva (Next Generation Sequencing [NGS]), detectando una variante patogénica del gen TRPS1 (c.333delC, p.Ser112Profs*7, NM_014112), en heterocigosis, que confirmaba el diagnóstico de TRPS tipo I.
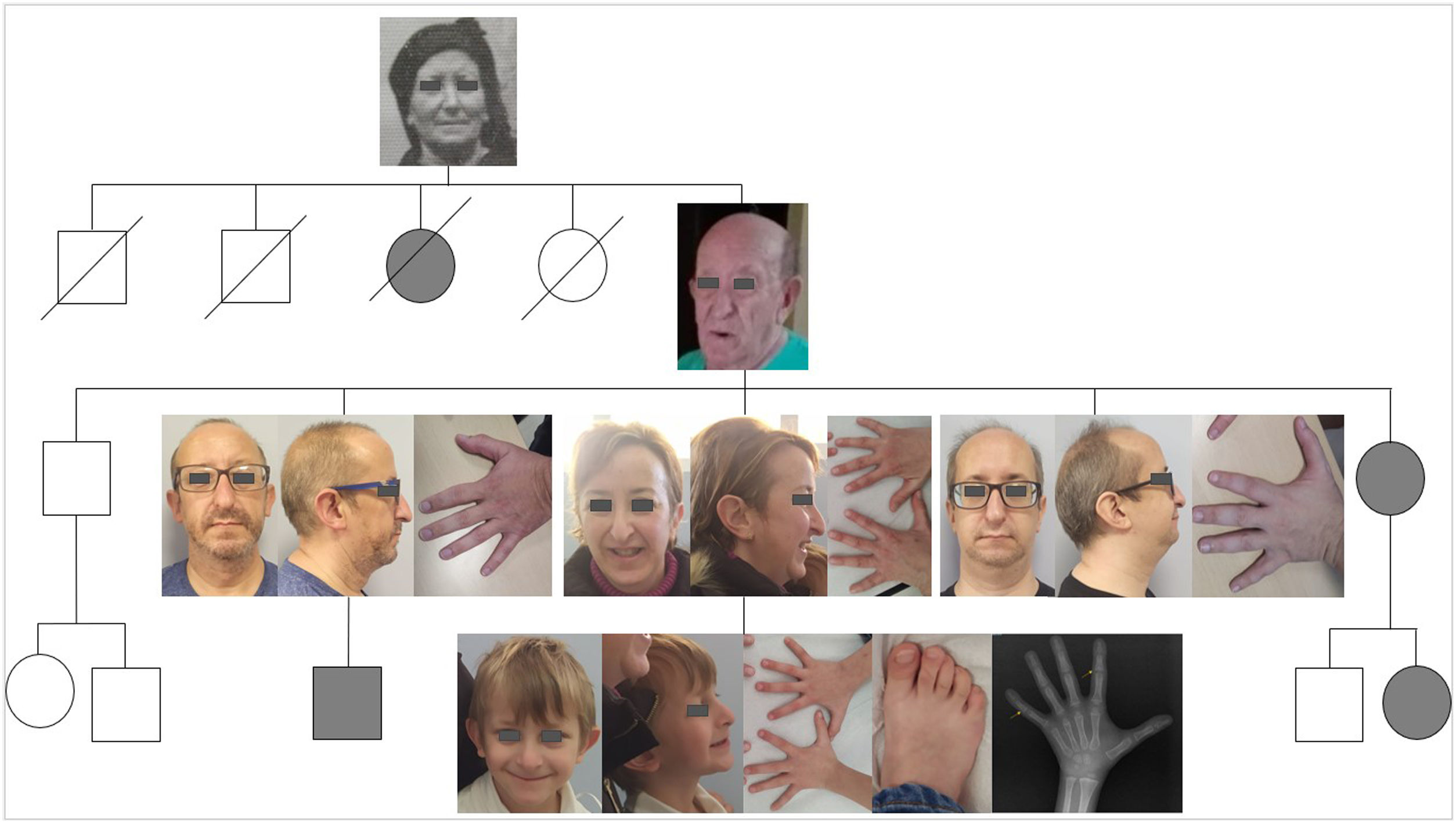
Realizamos un estudio de la familia materna, en el que participaron la madre y 3 tíos varones (fig. 1). En la tabla 1 mostramos los antecedentes, exploración y genética de ellos. Tanto la madre como dos de los tíos tenían fenotipo compatible a priori, detectándose en ellos la misma variante patogénica de TRPS1 que en el niño, mientras que el tío con pocos rasgos compatibles no la presentaba. Podemos ver una variabilidad fenotípica intrafamiliar (también descrita en la literatura2), aunque comparten, en mayor o menor medida, alteraciones en el pelo y las uñas, rasgos faciales característicos y alteraciones esqueléticas.
Antecedentes personales, exploración física y estudio genético de los miembros de la familia con TRPS
| Caso índice | Madre | Primer tío | Segundo tío | Tercer tío | |
|---|---|---|---|---|---|
| Antecedentes personales | Retraso global del desarrollo leve;Hipertrofia adenoidea | Endometriosis moderada;Portadora retinosis pigmentaria | Hernia de hiato;Hipertensión arterial | Fracturas de fémur y tibia derechas intervenidas | Septoplastia por desviación septal |
| Nivel de estudios | Primaria | EGB | EGB y FP | EGB y FP | EGB y administrativo |
| Cabello y pestañas escasas | Sí | Sí | Sí | Sí | Sí |
| Adelgazamiento lateral de las cejas | Sí | Sí | Sí | Sí | Sí |
| Uñas frágiles o blanquecinas | Sí | Sí | No | No | Sí |
| Cara triangular | Sí | Sí | Sí | Sí | Sí |
| Punta nasal bulbosa | Sí | Sí | No | Sí | Sí |
| Filtrum largo y plano | Sí | Sí | No | Sí | Sí |
| Labio superior fino | Sí | Sí | No | Sí | Sí |
| Pabellones auriculares prominentes | Sí | Sí | No | Sí | Sí |
| Abombamiento frontal | Sí | Sí | No | Sí | Sí |
| Micrognatia | No | No | No | Sí | Sí |
| Clinodactilia del meñique | Sí | Sí | No | Sí | No |
| Talla baja(≤−2 DE) | Sí (−2,68 DE) | Sí (−2,83 DE) | No (−1,96 DE) | Sí (−3,23 DE) | No (−1,54 DE) |
| Hiperlordosis o escoliosis | No | Sí | No | Sí | No |
| Hipotonía o hiperlaxitud | Sí | No | No | Sí | No |
| Otros | Sindactilia 2°-3er dedos de pies | Leve dismetría de miembros | |||
| Variante patogénica TRPS1 | Sí | Sí | No | Sí | Sí |
Por tanto, el diagnóstico de TRPS está al alcance del pediatra pues se puede realizar con los datos recogidos en la anamnesis y exploración física, con el apoyo de la radiografía simple, en la que podemos observar epífisis de las falanges en forma de cono, características de esta entidad; y con el estudio genético dirigido se puede confirmar la presencia de alteraciones en el gen TRPS1.
La mayor parte de la morbilidad en estos pacientes está determinada por la afectación osteoarticular, en forma osteoartritis de inicio precoz (sobre todo caderas, pero también otras articulaciones grandes y manos), con alteración de la movilidad, dolor articular y desviación de las falanges; e incluso aumento de la frecuencia de fracturas, por lo que el diagnóstico del síndrome puede adelantar el manejo de estas complicaciones. No es frecuente la asociación con discapacidad intelectual en general y si esta existe suele ser leve, excepto en el TRPS II en que suele ser más común1,2.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Presentación previa: 36 Congreso Nacional SEPEAP. Alicante, 20-22 de octubre de 2022.
