




Trichorhinophalangeal syndrome (TRPS) is an infrequent autosomal dominant syndrome with a high penetrance and variable expressivity caused by a change in the TRPS1 gene. Its clinical presentation is characterised by abnormalities of the hair (sparse scalp hair, lateral thinning of the brows) and nails (ungual dystrophy), mild facial dysmorphism (bulbous tip of the nose, long and flat philtrum, thin upper lip and protruding ears) and skeletal abnormalities (short stature, brachydactyly, phalangeal deviation, cone-shaped epiphyses at the phalanges, hip dysplasia and osteopenia). It is classified into two types: TRPS I (OMIM # 190350), caused by pathogenic variants of the TRPS1 gene; and TRPS II (OMIM # 150230), caused by the deletion of contiguous genes in chromosome 8 (including TRPS1 and EXT1), which is also associated with osteochondroma and intellectual disability.1–3
In this article, we present the cases of several members of a family who received a diagnosis of TRPS stemming from the evaluation of a boy aged 6 years for growth delay and certain dysmorphic features. In the index case, the main findings of the physical examination were a height of 108.8 cm (height z score, −2.68), a body mass index (BMI) of 13.52 kg/m2 (10th percentile), with normal body proportions and a height velocity 5.1 cm/year. He had fair, thin and sparse hair, thinning of the tail of the eyebrows and brittle nails. Other salient features were the triangular shape of the face, bulging forehead, long and flat philtrum, thin upper lip and large and retroverted ears. In the extremities, there was clinodactyly of the fifth toe and abnormally proximal position of the first toe joints. The patient also exhibited joint hypermobility, with flexible flatfoot and a scoliotic posture in the absence of abnormal vertebral rotation. He had been born to term with a birth weight of 2690 g (7th percentile) and a birth length of 49 cm (28th percentile) and had a personal history of mild global developmental delay and adenoidectomy for treatment of adenoid hypertrophy.
There was no history of consanguinity in the family. The salient findings of the family history were maternal short stature (147.2 cm; <1st percentile; height z score, −2.83) associated with a phenotype similar to that of the patient. The father’s height was normal (175 cm; 36th percentile). On interviewing the mother, she reported that there were several members of her family with short stature and similar facial features.
Hormone levels (IGF1, IGFBP3 and thyroid hormones) were normal, and the patient tested negative for markers of coeliac disease. His bone age was delayed by 3 years and the epiphyses of several phalanges were cone-shaped (Fig. 1). Genetic testing with next generation sequencing (NGS) panel of genes involved in bone dysplasias detected a heterozygous pathogenic variant in the TRPS1 gene (c.333delC, p.Ser112Profs*7, NM_014112), confirming the diagnosis of TRPS type I.
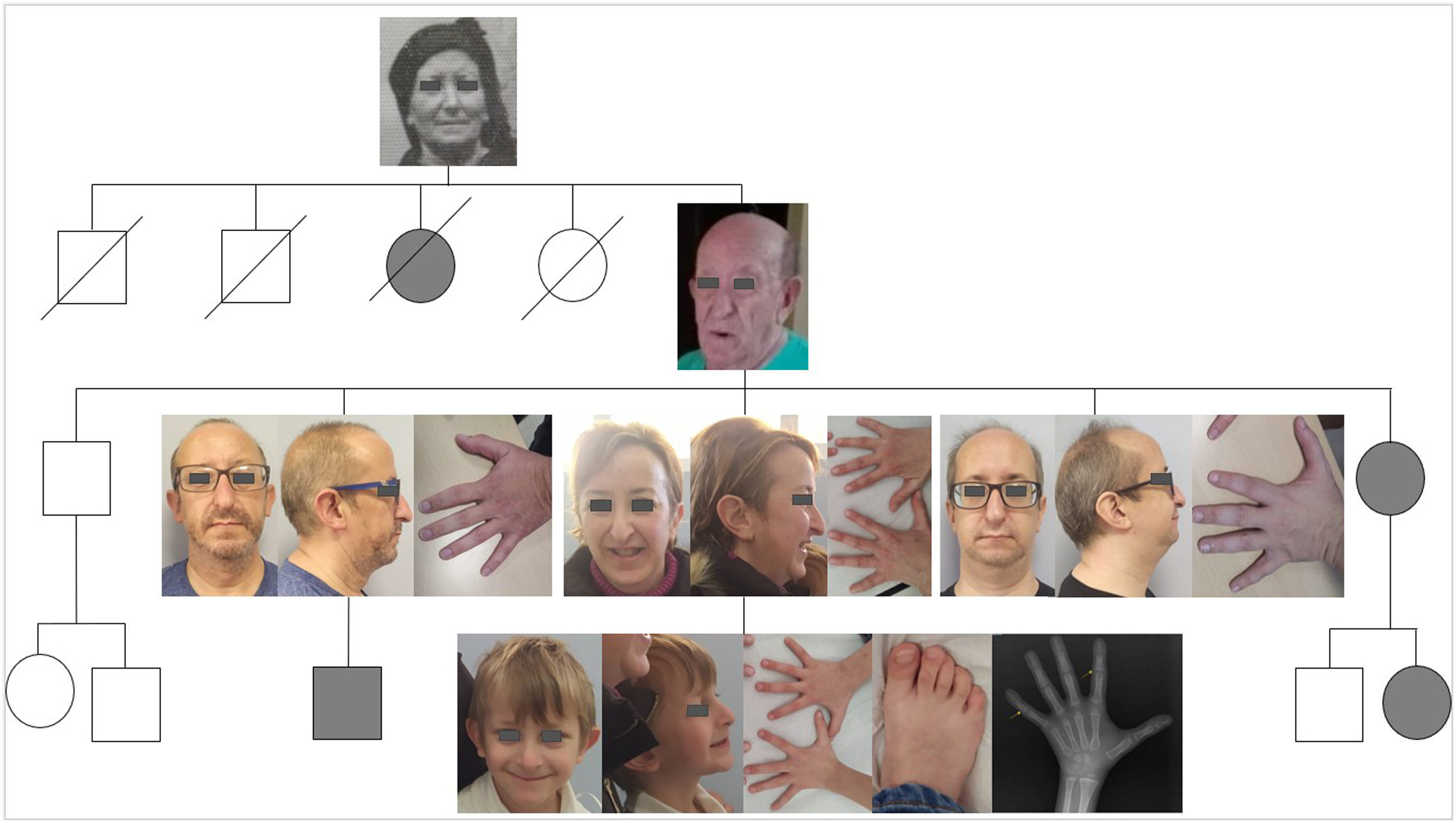
We performed cascade testing in the mother’s family, with participation of the mother and 3 male uncles (Fig. 1). Table 1 presents the findings of the history-taking, examination and genetic testing of the family. Both the mother and two of the uncles had a compatible phenotype, and all were found to have the same pathogenic variant of the TRPS1 gene as the boy, which was not present in the uncle with few compatible features. We observed phenotypic variability within the family (previously reported in the literature2), although they shared, to a varying extent, hair and nail abnormalities, the characteristic facial features and the skeletal changes.
Personal history and findings of physical examination and genetic testing in members of the family with TRPS.
| Personal history | Index case | Mother | Uncle 1 | Uncle 2 | Uncle 3 |
|---|---|---|---|---|---|
| Mild global developmental delay; adenoid hypertrophy | Moderate endometriosis; retinitis pigmentosa carrier | Hiatal hernia; hypertension | Operated right femur and tibia fractures | Septoplasty for deviated septum | |
| Educational attainment | Primary education | EGBa | EGBa and VE | EGBa and VE | EGBa and administrative training |
| Sparse hair and lashes | Yes | Yes | Yes | Yes | Yes |
| Thinning of brow tails | Yes | Yes | Yes | Yes | Yes |
| Brittle or pale nails | Yes | Yes | No | No | Yes |
| Triangular facies | Yes | Yes | Yes | Yes | Yes |
| Bulbous nose | Yes | Yes | No | Yes | Yes |
| Long and flat philtrum | Yes | Yes | No | Yes | Yes |
| Thin upper lip | Yes | Yes | No | Yes | Yes |
| Protruding ears | Yes | Yes | No | Yes | Yes |
| Bulging forehead | Yes | Yes | No | Yes | Yes |
| Micrognathia | No | No | No | Yes | Yes |
| Clinodactyly of fifth finger and toe | Yes | Yes | No | Yes | No |
| Short stature (height z ≤ −2) | Yes (z −2.68) | Yes (z −2.83) | No (z −1.96) | Yes (z −3.23) | No (z −1.54) |
| Hyperlordosis or scoliosis | No | Yes | No | Yes | No |
| Hypotonia or hypermobility | Yes | No | No | Yes | No |
| Other | Syndactyly (2nd and 3rd toes) | Mild limb length discrepancy | |||
| Pathogenic TRPS1 variant | Yes | Yes | No | Yes | Yes |
VE, vocational education.
Thus, the diagnosis of TRPS is feasible for paediatricians, as it can be reached with the information obtained in the history-taking and physical examination and supported by plain radiography, which allows visualization of the cone-shaped phalangeal epiphyses characteristic of this disease, after which targeted gene sequencing can confirm the presence of changes in the TRPS1 gene.
Most of the morbidity in these patients is determined by osteoarticular changes, in the form of osteoarthritis with an early onset (chiefly involving the hips, but also other large joints and the hands), abnormalities in joint mobility, articular pain and phalangeal deviation, so early diagnosis of the syndrome makes it possible to manage these complications from an earlier stage. The syndrome is not usually associated with intellectual disability and, when the latter is present, it tends to be mild, except in patients with TRPS type II, in whom it is more common.1,2
FundingThis research did not receive any external funding.
Previous meeting: this report was presented at the 36th National Congress of the Sociedad Española de Pediatría Extrahospitalaria y Atención Primaria (SEPEAP); October 20–22, 2022; Alicante, Spain.
